 | Информационная система | |
ГОСУДАРСТВЕННОЕ
САНИТАРНО-ЭПИДЕМИОЛОГИЧЕСКОЕ
НОРМИРОВАНИЕ РОССИЙСКОЙ ФЕДЕРАЦИИ
4.1. МЕТОДЫ КОНТРОЛЯ. ХИМИЧЕСКИЕ ФАКТОРЫ
МЕТОДИКА ВЫПОЛНЕНИЯ
ИЗМЕРЕНИЙ МАССОВОЙ ДОЛИ
СВИНЦА И КАДМИЯ В ПИЩЕВЫХ ПРОДУКТАХ И
ПРОДОВОЛЬСТВЕННОМ СЫРЬЕ МЕТОДОМ
ЭЛЕКТРОТЕРМИЧЕСКОЙ АТОМНО-АБСОРБЦИОННОЙ
СПЕКТРОМЕТРИИ
МЕТОДИЧЕСКИЕ УКАЗАНИЯ
МУК 4.1.986-00
Минздрав России
Москва
1. Разработаны Федеральным центром Госсанэпиднадзора Минздрава России (Брагина И.В., Ермаченко Л.А., Ерохина С.И.) и ООО «КОРТЭК» (Садагов Ю.М., Егорова И.А.).
2. Рекомендованы к утверждению Комиссией по госсанэпиднормированию при Минздраве России.
3. Утверждены и введены в действие Главным государственным санитарным врачом Российской Федерации, Первым заместителем министра здравоохранения Российской Федерации.
4. Введены впервые.
СОДЕРЖАНИЕ
УТВЕРЖДАЮ
Главный государственный санитарный
врач Российской Федерации
Г.Г. Онищенко
13 октября 2000 г.
Дата введения: 1 января 2001 г.
4.1. МЕТОДЫ КОНТРОЛЯ. ХИМИЧЕСКИЕ ФАКТОРЫ
Методика выполнения
измерений массовой доли свинца и
кадмия в пищевых продуктах и продовольственном
сырье методом электротермической атомно-
абсорбционной спектрометрии
Методические
указания
МУК 4.1.986-00
1. Назначение и область применения
Настоящие методические указания устанавливают метод атомно-абсорбционной спектрометрии с атомизацией в графитовой печи для определения содержания свинца и кадмия в пищевых продуктах и продовольственном сырье и предназначены для проведения лабораторных исследований безопасности пищевой продукции учреждениями госсанэпидслужбы России, а также для предприятий и учреждений, осуществляющих контроль качества и исследование пищевых продуктов и продовольственного сырья в соответствии с СанПиН 2.3.2.560-96 «Гигиенические требования к качеству и безопасности продовольственного сырья и пищевых продуктов» и аккредитованных в установленном порядке.
2. Метрологические характеристики методики
Приведенные в настоящих методических указаниях оптимальные аппаратурные параметры и условия проведения измерений относятся к атомно-абсорбционному спектрометру с электротермическим атомизатором «Квант-Z.ЭТА». При использовании спектрометров других марок эти параметры должны быть согласованы с требованиями Руководства по эксплуатации фирмы-изготовителя.
Диапазон определения массовых долей элементов в пищевых продуктах приведен в табл. 2.1.
Диапазон определения массовых долей элементов
|
Диапазон определения массовой доли элемента в продукте, мг/кг |
|
|
Кадмий |
0,01 - 2,0 |
|
Свинец |
0,02 - 10,0 |
Диапазон определяемых массовых концентраций элементов в растворах для анализа, полученных в результате минерализации образцов пищевых продуктов и пищевого сырья и последующего разбавления минерализата, приведен в табл. 2.2.
Диапазон определяемых массовых концентраций элементов в растворах для анализа
|
Диапазон массовых концентраций элемента в растворе, мкг/дм3 |
|
|
Кадмий |
0,1 - 2,0 |
|
Свинец |
2,0 - 40,0 |
Настоящая методика с вероятностью Р = 0,95 обеспечивает получение результатов анализа с погрешностью, не превышающей значений, приведенных в табл. 2.3.
Характеристики погрешности измерений
|
Диапазон содержаний, мг/кг |
Сходимость dотн, % |
Воспроизводимость Dотн, % |
Границы относительной погрешности d, % |
|
|
Кадмий |
0,01 - 2,0 |
22 |
36 |
± 30 |
|
Свинец |
0,02 - 1,0 |
28 |
42 |
± 35 |
3. Метод измерений
Метод основан на измерении оптической плотности атомного пара определяемого элемента, образующегося в результате электротермической атомизации минерализата пищевого продукта в графитовой печи атомно-абсорбционного спектрометра, на резонансной спектральной линии.
4. Средства измерений, вспомогательные устройства, материалы и реактивы
4.1. Средства измерений
|
4.1.1. Атомно-абсорбционный (АА) спектрометр с электротермическим атомизатором «Квант-Z.ЭТА», с корректором неселективного (фонового) поглощения на основе эффекта Зеемана, или другой, с аналогичными техническими и метрологическими характеристиками, укомплектованный лампами полого катода (ЛПК) на кадмий и свинец |
ТУ 4434-009-2990357-95 |
|
4.1.2. Весы лабораторные общего назначения, 2-го класса точности с наибольшим пределом взвешивания 200 г |
|
|
4.1.3. Набор гирь Г-2-200 |
|
|
4.1.4. Государственные стандартные образцы (ГСО) состава растворов ионов кадмия и свинца с массовой концентрацией элементов в 1,0 мг/см3 и относительной погрешностью концентрации не более 1 % |
|
|
4.1.5. Пипетки мерные 2-1-2-1 или 1-1-2-1, 2-1-2-2 или 1-1-2-2, 1-2-2-5 и 1-2-2-10 |
ГОСТ 29927 |
|
4.1.6. Одноканальные пипетки переменного объема 0,5 - 10 мкл и 200 - 1000 мкл с погрешностью дозирования не более 2 % или аналогичные пипетки с погрешностью дозирования не более 2 % |
|
|
4.1.7. Колбы мерные 2-25-2, 2-50-2, 2-100-2, 2-200-2 и 2-1000-2 |
|
|
4.1.8. Цилиндры 1-5, 1-10, 1-25, 1-50 и 1-500 |
Допускается использование других средств измерений с метрологическими характеристиками не хуже указанных выше.
4.2. Вспомогательные устройства и материалы
|
|
|
|
4.2.3. Автоклавы аналитические НПВФ «Анкон-АТ-2» с реакционной камерой, вместимостью 150 см3 |
ТУ 48-0572-31-259 |
|
4.2.4. Насос водоструйный или насос Камовского |
|
|
4.2.5. Щипцы тигельные |
|
|
4.2.6. Электроплитка бытовая или горелка газовая |
|
|
4.2.7. Баня водяная |
|
|
4.2.8. Чашки или тигли кварцевые №№ 2 - 4 или |
|
|
чашки (тигли) фарфоровые |
|
|
4.2.9. Стекла часовые или чашки Петри для накрывания тиглей (чашек) |
|
|
4.2.10. Воронки лабораторные |
|
|
4.2.11. Фильтры обезволенные с синей лентой, диаметром 7 - 10 см |
ТУ 6-09-1678 |
|
4.2.12. Стаканы В-1-50, В-1-100 или В-1-150 |
|
|
4.2.13. Колбы Кьельдаля 2-50-29, 2-100-29, 2-25-29 или колбы плоскодонные П-2-250-34 ТХС |
|
|
4.2.14. Стаканчики для взвешивания СВ-14/8 |
|
|
4.2.15. Колбы Кн-2-1000-29 или Кн-2-1000-34 |
|
|
4.2.16. Колбы конические, вместимостью 100 см3 |
|
|
4.2.17. Колбы Кн-1-250-29/32 ТСХ или П-1-250-29/32 ТС |
|
|
4.2.18. Холодильник ХIII-1-200-29/32 ТС |
|
|
4.2.20. Пробирки для микропроб типа «Эппендорф», вместимостью 1,5 - 2,0 см3 |
|
|
4.2.21. Шарики стеклянные, используемые для обеспечения равномерности кипения |
|
|
4.2.22. Синтетическое моющее средство (СМС) для мытья лабораторной посуды (2 г СМС растворить в 1000 см3 воды) |
|
4.3. Реактивы
|
4.3.1. Кислота азотная особой чистоты или очищенная методом перегонки |
|
|
4.3.2. Кислота соляная, особой чистоты |
|
|
4.3.3. Кислота серная |
|
|
4.3.4. Палладий металлический, порошок (99,94 %) |
|
|
4.3.5. Вода дистиллированная |
|
|
4.3.6. Вода бидистиллированная или деионизованная |
|
|
4.3.7. Водорода пероксид (пергидроль), х. ч. |
|
|
4.3.8. Аргон газообразный высокой чистоты |
Допускается применение других реактивов с техническими и метрологическими характеристиками не хуже указанных.
5. Требования безопасности
5.1. При выполнении анализов необходимо соблюдать требования техники безопасности при работе с химическими реактивами по ГОСТу 12.1.007.
5.2. Помещение, в котором проводят измерения, должно быть оборудовано общей приточно-вытяжной вентиляцией.
5.3. Над модулем атомизатора спектрометра должен быть установлен вытяжной зонт согласно техническому описанию и инструкции по эксплуатации ГКНЖ.15.00.000 ТО п. 8.5.
5.4. Не допускается эксплуатация спектрометра без заземления, а также использование для заземления нулевой фазы электропитания.
5.5. Электробезопасность при работе с электроустановками по ГОСТу 12.2.007.
5.6. Организация обучения работающих безопасности труда по ГОСТу 12.0.004.
5.7. Помещение лаборатории должно соответствовать требованиям пожарной безопасности по ГОСТу 12.1.004 и иметь средства пожаротушения по ГОСТу 12.4.009.
5.8. Содержание вредных веществ в воздухе рабочей зоны не должно превышать норм, установленных ГН 2.2.5.686-98 «Предельно допустимые концентрации (ПДК) вредных веществ в воздухе рабочей зоны».
5.9. При эксплуатации сжатых газов следует соблюдать «Правила устройства и безопасной эксплуатации сосудов, работающих под давлением» (ПБ 10-115-96), Госгортехнадзор России, ГОСТ 12.2.085.
6. Требования к квалификации персонала
6.1. К работе на атомно-абсорбционном спектрометре допускается персонал, прошедший соответствующий курс подготовки.
6.2. К работе по подготовке проб пищевых продуктов допускается персонал, имеющий навыки работы в химической лаборатории и прошедший обучение работе с аналитическими автоклавами (при проведении автоклавной пробоподготовки).
7. Условия выполнения измерений
При выполнении измерений необходимо соблюдать следующие условия.
Температура окружающего воздуха, °С (20±10)
Атмосферное давление, кПа (101±4)
(760±30) мм рт. ст.
Относительная влажность воздуха, % (65±15)
Напряжение питания сети, В 220 (205 - 230)
Частота питающей сети, Гц (50±0,5)
8. Подготовка к выполнению измерений
8.1. Настройка спектрометра
Включают и настраивают спектрометр согласно техническому описанию и инструкции по эксплуатации. Основные инструментальные параметры определения свинца и кадмия на спектрометре « Квант-Z.ЭТА» приведены в табл. 8.1.
Оптимальные аппаратурные параметры спектрометра «КВАНТ-Z.ЭТА»
|
Источник излучения |
Длина волны, нм |
Ток ЛПК, мА |
Ширина щели монохроматора, мм |
|
|
Кадмий |
ЛПК |
228,8 |
5 |
0,5 |
|
Свинец |
ЛПК |
283,3 |
10 |
0,25 |
Параметры программ нагрева графитовой печи прибора «Квант-Z.ЭТА» приведены в табл. 8.2.
Программы нагрева графитовой печи
|
Время, с |
Температура, °С |
Поток аргона |
|||
|
нарастание |
выдержка |
кадмий |
свинец |
||
|
Испарение |
9 |
9 |
50* |
Открыт |
|
|
Пиролиз 1 |
9 |
5 |
60* |
Открыт |
|
|
Пиролиз 2 |
9 |
15** |
500 |
800 |
Открыт |
|
Пиролиз 3 |
0 |
1 |
500 |
800 |
Закрыт |
|
Атомизация |
0 |
0,6 |
1800 |
2000 |
Закрыт |
|
Очистка |
0 |
2 |
2600 |
Открыт |
|
|
* Температура стадии «испарение» может изменяться на ±10 °С для разных экземпляров спектрометра. ** Время выдержки стадии «пиролиз 2» может быть увеличено на 5 - 10 с, если амплитудные значения фоновой абсорбционности превышают 1,2 Б. |
|||||
8.2. Приготовление растворов
Для приготовления растворов используют бидистиллированную или деионизованную воду.
8.2.1. Фоновый раствор азотной кислоты с концентрацией 0,03 моль/дм3. В мерную колбу, вместимостью 1000 см3, помещают 2 см3 азотной кислоты (п. 4.3.1) и доводят до метки бидистиллированной водой. Полученный раствор используют для разбавления проб и приготовления градуировочных растворов.
8.2.2. Раствор палладия с массовой концентрацией 10 мг/см3. Навеску 1,00 г металлического палладия помещают в термостойкий стакан, вместимостью 100 см3, добавляют 5 см3 концентрированной азотной кислоты и нагревают на электроплитке с закрытой спиралью до полного растворения металла. Раствор охлаждают, фильтруют в мерную колбу вместимостью 100 см3, доводят объем до метки фоновым раствором азотной кислоты (п. 8.2.1) и перемешивают. Концентрация палладия в приготовленном растворе равна 10 мг/см3. Допускается использовать коммерческий палладиевый модификатор для атомно-абсорбционной спектрометрии с концентрацией палладия 10 г/дм3 производства фирмы Меrck.
8.2.3. Фоновый раствор палладия с массовой концентрацией 20 мг/дм3. В мерную колбу, вместимостью 1000 см3, помещают 2 см3 раствора палладия (п. 8.2.2), доводят до метки фоновым раствором азотной кислоты (п. 8.2.1) и перемешивают.
8.2.4. Исходный раствор (А) с массовой концентрацией кадмия, свинца 10 мг/дм3. Вскрывают ампулу ГСО соответствующего элемента (п. 4.1.4) с концентрацией 1 мг/см3, выливают в сухой стакан, с помощью пипетки, вместимостью 1 см3, отбирают 1 см3 раствора ГСО, переносят в мерную колбу, вместимостью 100 см3, и доводят до метки фоновым раствором азотной кислоты (п. 8.2.1). В отсутствие ГСО допускается использование стандартных растворов элементов, приготовленных согласно ГОСТу 4212. Срок хранения раствора - 6 месяцев.
8.2.5. Раствор (Б) с массовой концентрацией кадмия, свинца 200 мкг/дм3. Отбирают пипеткой 2 см3 раствора А (п. 8.2.4), переносят в мерную колбу, вместимостью 100 см3, и доводят до метки фоновым раствором азотной кислоты (п. 8.2.1). Срок хранения раствора 10 дней.
8.2.6. Раствор (В) с массовой концентрацией кадмия 10 мкг/дм3. Отбирают пипеткой 5 см3 раствора Б кадмия (п. 8.2.5), переносят в мерную колбу, вместимостью 100 см3, доводят до метки фоновым раствором азотной кислоты (п. 8.2.1). Раствор готовят в день анализа.
8.3. Градуировка спектрометра
8.3.1. Градуировку выполняют согласно техническому описанию и инструкции по эксплуатации АА спектрометра.
Число измерений каждого градуировочного раствора - 3. Относительное среднеквадратическое отклонение результатов параллельных измерений каждого градуировочного раствора не должно превышать 10 %.
8.3.2. Градуировочные растворы элементов готовят в день проведения анализа в мерных колбах, вместимостью 25, 50 и 100 см3. Растворы в колбах доводят до метки фоновым раствором палладия (п. 8.2.3).
8.3.3. Приготовление растворов для градуировки проводят согласно табл. 8.3.
8.3.4. В качестве «нулевого» раствора при приготовлении градуировочных растворов элементов и при проведении градуировки используют фоновый раствор палладия с массовой концентрацией 20 мг/дм3 (п. 8.2.3).
Приготовление градуировочных растворов свинца и кадмия
|
Массовая концентрация в градуировочном растворе, мкг/дм3 |
Объем мерной колбы, см3 |
Аликвоты разбавляемых растворов, помещаемых в мерную колбу |
||
|
кадмий |
свинец |
|||
|
1 |
0,1 |
2 |
100 |
|
|
2 |
0,5 |
10 |
100 |
|
|
3 |
1,0 |
20 |
100 |
|
|
4 |
2,0 |
40 |
50 |
|
Типичные градуировочные графики кадмия и свинца для АА спектрометра «Квант-Z.ЭТА» приведены на рис. 1 и 2, соответственно.
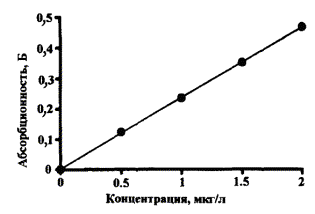
Рис. 1. Градуировочный график для определения кадмия.
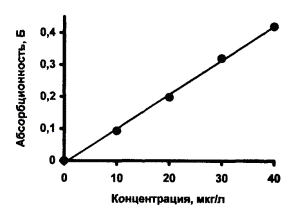
Рис. 2. Градуировочный график для определения свинца.
8.3.5. Стабильность градуировочной характеристики контролируют не реже, чем через каждые 20 анализируемых проб. Образцами для контроля стабильности градуировочных характеристик являются градуировочные растворы №№ 2 или 3, согласно табл. 8.3. Градуировочную характеристику считают стабильной и используют в дальнейших измерениях, если выполняется следующее условие:
![]() , где (1)
, где (1)
С0 - действительное значение массовой концентрации элемента в градуировочном растворе, мкг/дм3;
С - измеренное значение массовой концентрации элемента в этом же градуировочном растворе, мкг/дм3;
d - значение относительной погрешности, приведенное в табл. 2.3.
В случае невыполнения условия (1) проводят коррекцию имеющейся градуировки или проводят повторную градуировку.
9. Выполнение измерений
9.1. Отбор проб
9.1.1. Отбор проб проводят в соответствии с нормативно-технической документацией на конкретный вид анализируемой продукции.
9.1.2. Для каждой пробы выполняют 2 параллельных определения.
9.2. Подготовка посуды
Используемую для анализа посуду промывают раствором СМС (п. 4.2.19), водопроводной водой, ополаскивают дистиллированной водой и тщательно промывают раствором азотной кислоты с концентрацией 1 моль/дм3. Непосредственно перед использованием посуду ополаскивают фоновым раствором азотной кислоты (п. 8.2.1).
9.3. Подготовка проб к анализу (минерализация проб)
9.3.1. Сухая минерализация
Подготовку проб методом сухой минерализации проводят согласно ГОСТу 26929 [1].
9.3.1.1. Способ сухой минерализации применяют для всех видов сырья и продуктов, кроме продуктов с содержанием жира больше 60 %, а также животных и растительных жиров и масел.
9.3.1.2. В фарфоровый или кварцевый тигель (чашку) берут навеску продукта массой 2 - 5 г из подготовленной к испытаниям пробы. Необходимый объем жидкого продукта отмеряют пипеткой.
9.3.1.3. Продукты, содержащие углекислый газ (пиво, шипучие и игристые вина, минеральные воды, газированные напитки и соки), освобождают от него.
При анализе пива колбу, вместимостью 1000 см3, на треть заполняют пивом (температура продукта должна быть комнатной), закрывают пробкой с отверстием, в которое вставлена трубка, и встряхивают в течение 20 - 30 мин.
При анализе вина, минеральной воды, газированных соков и напитков в пробе продукта, помещенного в колбе с тубусом, создают вакуум при помощи водоструйного или масляного насоса в течение 2 - 3 мин до исчезновения пены и появления больших пузырей на поверхности жидкости.
9.3.1.4. При содержании в продукте до 20 % влаги тигель (чашку) с навеской помещают на электроплитку и проводят осторожно обугливание, не допуская сильного дымления. После прекращения выделения дыма тигель (чашку) помещают в электропечь, отрегулированную ранее на температуру около 250 °С.
При содержании влаги в продукте от 20 до 80 % тигель (чашку) с навеской помещают на кипящую водяную баню или в сушильный шкаф (доводя его температуру до 150 °С), или на электрическую плитку и удаляют влагу. Затем осторожно обугливают содержимое тигля (чашки) на газовой горелке или электрической плитке до прекращения выделения дыма, не допуская сильного дымления, воспламенения и выбросов. Тигель (чашку) помещают в электропечь, отрегулированную ранее на температуру около 250 °С, а продукцию, содержащую более 20 % сахаров, помещают в электропечь, отрегулированную ранее на температуру около 150 °С.
При содержании в продукте влаги более 80 % навеску в тигле (чашке) обрабатывают следующим образом:
· винодельческие продукты упаривают досуха на водяной бане и помещают в электропечь;
· пиво, минеральную воду, безалкогольные напитки и плодоовощные соки и напитки на электроплитке упаривают досуха и проводят обугливание до прекращения выделения дыма, затем помещают в электропечь, отрегулированную на температуру около 150 °С;
· в навеску жидких молочных продуктов (молока, кисломолочных продуктов и молочных консервов) добавляют раствор азотной кислоты из расчета 1 см3 на 50 г продукта, перемешивают, помещают на электроплитку и осторожно проводят обугливание до прекращения выделения дыма, затем тигель (чашку) помещают в электропечь, отрегулированную ранее на температуру около 250 °С.
9.3.1.5. После окончания обугливания минерализацию проб проводят в электропечи, постепенно повышая температуру до 450 °С:
250 °С - 30 мин;
300 °С - 30 мин;
350 °С - 30 мин;
400 °С - 30 мин;
450 °С - 1 - 3 ч до получения серой золы.
Допускается минерализация зерна и зернопродуктов при температуре до 500 °С.
9.3.1.6. Тигель с золой вынимают из электропечи, охлаждают до комнатной температуры, золу смачивают раствором азотной кислоты (1:1), раствор выпаривают досуха на плитке. После охлаждения тигель с навеской снова помещают в электропечь, нагретую до температуры 250 °С. Постепенно доводят температуру до 400 °С и выдерживают 30 мин. Указанный цикл повторяют несколько раз. Минерализацию считают законченной, когда зола станет белой или слегка окрашенной, без обугленных частиц. При наличии обугленных частиц повторяют обработку раствором азотной кислоты (1:1).
9.3.1.7. В тигель (чашку) с золой прибавляют 1 см3 раствора азотной кислоты (1:1) и нагревают на водяной бане до растворения солей. Затем раствор упаривают до влажных солей, добавляют 5 см3 фонового раствора азотной кислоты (п. 8.2.1), слегка нагревают на водяной бане до полного растворения солей, охлаждают и количественно переносят в мерную колбу на 25 см3. Доводят до метки фоновым раствором азотной кислоты.
9.3.1.8. Одновременно с партией проб готовят 2 контрольных («холостых») опыта для контроля чистоты посуды и реактивов, добавляя в тигель все реактивы, но без навески пробы, и точно повторяя все условия (количество реактивов, температуру, время нагрева), в которых выполняется минерализация пробы.
9.3.2. Кислотная минерализация
Подготовку проб методом мокрой минерализации (открытый способ) проводят согласно ГОСТу 26929.
9.3.2.1. Способ основан на полном разрушении органических веществ пробы продукта при нагревании с кислотами-окислителями и перекисью водорода и предназначен для всех видов сырья и продуктов, кроме сливочного масла и животных жиров.
9.3.2.2. Отбирают навеску продукта массой 2 - 5 г из подготовленной для испытания пробы.
9.3.2.2.1. Навеску жидких и пюреобразных продуктов взвешивают в стакане, переносят в колбу Кьельдаля или плоскодонную колбу, смывая стенки стакана 10 - 15 см3 дистиллированной воды. Допускается брать навеску непосредственно в плоскодонную колбу.
9.3.2.2.2. Навеску твердых и пастообразных продуктов берут на обеззоленный фильтр, заворачивают в него и стеклянной палочкой помещают на дно колбы Кьельдаля или плоскодонной колбы.
9.3.2.2.3. Навеску сухих продуктов помещают в колбу Кьельдаля, добавляют 15 см3 воды, перемешивают.
9.3.2.2.4. Желатин оставляют на 1 ч для набухания.
9.3.2.3. В колбу с навеской продукта, подготовленной к минерализации, вносят азотную кислоту из расчета 10 см3 на каждые 5 г продукта, на каждые 1 - 2,5 г консервированного молока и выдерживают не менее 15 мин. Можно оставить на ночь. Затем в колбу вносят 2 - 3 стеклянных шарика для равномерного кипения, закрывают грушевидной пробкой и начинают постепенно нагревать на электроплитке, упаривая содержимое колбы до объема 3 - 5 см3.
9.3.2.4. Колбы охлаждают, вносят 10 см3 азотной кислоты, содержимое упаривают до объема 5 см3, охлаждают. Эту процедуру повторяют 2 - 4 раза.
9.3.2.5. В колбу вносят 10 см3 азотной кислоты, 5 см3 серной кислоты, 4 см3 пероксида водорода из расчета на каждые 5 г продукта. Не допускается изменять последовательность внесения кислот, пероксид водорода всегда добавляется последним. Содержимое колбы упаривают до объема около 5 см3, не допуская образования коричневой окраски жидкости. При появлении коричневой окраски нагревание прекращают.
9.3.2.6. Колбу охлаждают до комнатной температуры, добавляют 5 см3 азотной кислоты и 2 см3 пероксида водорода и снова нагревают до появления белых паров серного ангидрида. Если при этом раствор не обесцветился, процедуру повторяют. Минерализацию считают законченной, если раствор после охлаждения остается бесцветным или бледно-желтым.
9.3.2.7. Для удаления остатков кислот в охлажденную колбу добавляют 10 см3 воды и кипятят 10 мин с момента выделения белых паров, затем охлаждают. Добавление воды и нагревание повторяют еще 2 раза.
9.3.2.8. После охлаждения полученный минерализат переносят в мерную колбу на 25 см3, доводят до метки дистиллированной водой и тщательно перемешивают.
9.3.2.9. Одновременно с партией проб проводят 2 контрольных опыта для контроля чистоты посуды и реактивов, добавляя в колбу реактивы в тех же количествах, что и в пробы, и точно повторяя все условия, в которых проводят минерализацию пробы.
9.3.3. Кислотная экстракция (неполная минерализация)
9.3.3.1. Способ основан на экстракции токсичных элементов из пробы продукта кипячением с разбавленной азотной кислотой и предназначен для растительного и сливочного масел, маргарина, пищевых жиров и сыров.
9.3.3.2. Экстракция проб продукта.
В термостойкую колбу с навеской продукта массой 5 - 10 г вносят цилиндром 40 см3 раствора азотной кислоты (1:2) по объему.
В колбу добавляют несколько стеклянных шариков, вставляют в нее холодильник, помещают на электроплитку, покрытую асбестом, и кипятят в течение 1,5 ч с момента закипания. Затем содержимое колбы медленно охлаждают до комнатной температуры, не вынимая холодильника.
Колбу с экстракционной смесью сливочного масла, жиров или маргарина с кислотой помещают в холодную водяную баню для затвердевания жира. Затвердевший жир прокалывают стеклянной палочкой, водный слой фильтруют через фильтр, смоченный раствором кислоты, используемой для экстракции, в кварцевую или фарфоровую чашку. Оставшийся в колбе жир расплавляют на водяной бане, добавляют 10 см3 раствора используемой кислоты, встряхивают, охлаждают, после охлаждения жир прокалывают и промывную жидкость сливают в тот же сосуд через тот же фильтр, затем фильтр промывают 5 - 7 см3 воды.
Экстракционную смесь растительного масла с кислотой переносят в делительную воронку. Колбу ополаскивают 10 см3 раствора используемой кислоты, который сливают в ту же воронку. После разделения слоев нижний водный слой сливают через фильтр, смоченный раствором используемой кислоты, в кварцевую или фарфоровую чашку, затем фильтр промывают 5 - 7 см3 воды.
Экстракционную смесь сыра с кислотой фильтруют через фильтр, смоченный раствором используемой кислоты, в кварцевую или фарфоровую чашку. Колбу ополаскивают 10 см3 раствора кислоты, который фильтруют через тот же фильтр, затем фильтр промывают 5 - 7 см3 воды.
9.3.3.3. Подготовка экстрактов для атомно-абсорбционного анализа.
Экстракционную смесь, полученную по п. 9.3.3.2, фильтруют в кварцевую или фарфоровую чашку. Жидкость осторожно выпаривают, а затем обугливают на электроплитке. Затем чашку помещают в электропечь и далее продолжают минерализацию по п. 9.3.1.5.
9.3.3.4. Параллельно в двух колбах проводят экстракцию и подготовку экстрактов к анализу добавляемых к навеске реактивов для контроля их чистоты.
9.3.4. Автоклавная минерализация
Процедуру минерализации проб в аналитических автоклавах проводят в соответствии с МУК 4.1.985-00 [2].
9.3.4.1. Метод основан на полной минерализации пробы смесью азотной кислоты и пероксида водорода в реакционной камере аналитического автоклава с резистивным нагревом.
9.3.4.2. Масса пробы для данного вида продукции не должна превышать величину, указанную в табл. 9.1.
Условия минерализации проб пищевой продукции и продовольственного сырья в автоклаве НПВФ «АНКОН-АТ-2» с реакционной камерой, вместимостью 150 см3
|
Масса навески, г |
Объем реактивов, см3 |
Экспозиция (комнатная температура) |
Температура и время нагрева (ч) |
Примечание |
|
|
1 |
2 |
3 |
4 |
5 |
6 |
|
1. Фрукты, овощи и продукты их переработки |
|||||
|
Свежие овощи, фрукты |
5,0 |
НNО3 - 6 Н2О2 - 1 |
- |
160 °С - 3 ч |
|
|
Листовые овощи: петрушка, укроп, салат |
3,0 |
НNО3 - 6 Н2О2 - 1 |
- |
160 °С - 3 ч |
|
|
Консервы ягодные |
3,0 |
НNО3 - 6 Н2О2 - 1 |
30 - 40 мин |
160 °С - 1 ч 180 °С - 2 ч |
|
|
Фрукты сушеные |
2,5 |
НNО3 - 7 Н2О2 - 1 |
50 - 60 мин |
160 °С - 1 ч 180 °С - 2 ч |
|
|
Орехи (грецкие, миндаль, фундук) |
2,0 |
НNО3 - 7 Н2О2 - 1 |
Заливают на ночь НNО3, утром добавляют Н2О2 |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
|
|
Грибы свежие |
3,0 |
НNО3 - 6 Н2О2 - 1 |
- |
160 °С - 1 ч 180 °С - 2 ч |
|
|
Грибы сушеные |
2,0 |
НNО3 - 6 Н2О2 - 1 |
20 - 30 мин |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
|
|
2. Мясо и мясопродукты |
|||||
|
Мясо (говядина, баранина, свинина, птица) |
2,5 |
НNО3 - 6 Н2О2 - 1 |
30 - 40 мин |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
Для жирных сортов - заливают на ночь НNО3, утром добавляют Н2О2 |
|
Колбасные изделия: колбасы вареные, сосиски, сардельки |
3,0 |
НNО3 - 6 Н2О2 - 1 |
30 - 40 мин |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
|
|
Колбасы сырокопченые, варено-копченые, полукопченые; рулеты |
2,5 |
НNО3 - 6 Н2О2 - 1 |
Заливают на ночь НNО3, утром добавляют Н2О2 |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
|
|
Субпродукты (печень, сердце, легкое, почки) |
3,0 |
НNО3 - 6 Н2О2 - 1 |
30 - 40 мин |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
|
|
Консервы мясные (паштет, тушенка в собственном соку, фарши) |
2,5 |
НNО3 - 6 Н2О2 - 1 |
30 - 40 мин |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
Для жирных сортов - заливают на ночь НNО3, утром добавляют Н2О2 |
|
3. Рыбные продукты |
|||||
|
Рыба живая, охлажденная, мороженная |
3,0 |
НNО3 - 6 Н2О2 - 1 |
30 - 40 мин |
160 °С - 1 ч 180 °С - 2 ч 200 °С - 1 ч |
Для жирных сортов - заливают на ночь НNО3, утром добавляют Н2О2 |
|
Рыба холодного и горячего копчения, жирная |
2,0 |
НNО3 - 7 Н2О2 - 1 |
Заливают на ночь НNО3, утром добавляют Н2О2 |
160 °С - 1 ч 180 °С - 2 ч 200 °С - 1 ч |
|
|
Беспозвоночные (креветки, ракообразные) |
3,0 |
НNО3 - 6 Н2О2 - 1 |
30 - 40 мин |
160 °С - 1 ч 180 °С - 2 ч 200 °С - 1 ч |
|
|
Консервы рыбные в собственном соку, в томате |
3,0 |
НNО3 - 6 Н2О2 - 1 |
30 - 40 мин |
160 °С - 1 ч 180 °С - 2 ч 200 °С - 1 ч |
- |
|
Консервы рыбные в масле |
2,0 |
НNО3 - 7 Н2О2 - 1 |
Заливают на ночь НNО3, утром добавляют Н2О2 |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
|
|
4. Зерно и продукты его переработки |
|||||
|
Рожь, пшеница, ячмень, овес, чечевица, соя, фасоль и др. |
2,0 |
НNО3 - 8 Н2О2 - 2 |
Заливают на ночь НNО3, утром добавляют Н2О2 |
160 °С - 1 ч 180 °С - 2 ч 200 °С - 1 ч |
Зерно измельчают, смачивают водой до получения однородной массы |
|
Подсолнечник (семена, жмых, шрот) |
1,5 |
||||
|
Продукты переработки зерна (мука, крупа, побочные продукты мукомольной промышленности) |
2,0 |
НNО3 - 8 Н2О2 - 2 |
Заливают на ночь НNО3, утром добавляют Н2О2 |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
Смачивают дистиллированной водой до получения однородной массы |
|
Хлеб, хлебобулочные изделия (булки, бублики, сушки, баранки, сухари, палочки) |
2,0 |
НNО3 - 6 Н2О2 - 1 |
20 - 30 мин |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
Бублики, баранки, сухари, соломку, сушки измельчают и смачивают дистиллированной водой |
|
Макаронные изделия |
2,0 |
НNО3 - 7 Н2О2 - 1 |
30 - 40 мин |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
Измельчают и смачивают дистиллированной водой |
|
5. Растения, корма растительного и животного происхождения |
|||||
|
Травяная мука, силос, сено, солома, зеленая масса |
1,5 |
НNО3 - 8 Н2О2 - 1 |
30 - 40 мин |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
Сухие продукты смачивают дистиллированной водой |
|
Корма животного происхождения (костная, рыбная мука) |
1,5 |
НNО3 - 6 Н2О2 - 1 |
30 - 40 мин |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
Смачивают дистиллированной водой |
|
Пряности, лекарственное сырье растительного и животного происхождения (сухое) |
1,5 |
НNО3 - 7 Н2О2 - 1 |
20 - 30 мин |
160 °С - 1 ч 180 °С - 2 ч 200 °С - 1 ч |
Смачивают дистиллированной водой |
|
6. Кондитерские изделия |
|||||
|
Печенье, вафли, галеты, пряники и др. |
2,0 |
НNО3 - 6 Н2О2 - 1 |
30 - 40 мин |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
Смачивают дистиллированной водой |
|
Торты, рулеты и пирожные с кремом |
2,0 |
НNО3 - 7 Н2О2 - 1 |
Заливают на ночь НNО3, утром добавляют Н2О2 |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
|
|
Пастила, мармелад |
3,0 |
НNО3 - 6 Н2О2 - 1 |
30 - 40 мин |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 1 ч |
|
|
Шоколад, конфеты |
2,0 |
НNО3 - 6 Н2О2 - 1 |
30 - 40 мин |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
Измельчают до частиц размером 1 мм |
|
Халва |
2,0 |
НNО3 - 6 Н2О2 - 1 |
30 - 40 мин |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
|
|
Сахар-рафинад, песок |
2,0 |
НNО3 - 8 Н2О2 - 1 |
30 - 40 мин |
160 °С - 1 ч 180 °С - 2 ч 200 °С - 1 ч |
Смачивают дистиллированной водой |
|
7. Пищевые концентраты |
|||||
|
Завтраки сухие |
2,0 |
НNО3 - 6 Н2О2 - 1 |
20 - 30 мин |
160 °С - 1 ч 180 °С - 2 ч |
Смачивают дистиллированной водой |
|
Соусы кулинарные порошкообразные, бульонные кубики |
2,0 |
НNО3 - 6 Н2О2 - 1 |
20 - 30 мин |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 1 ч |
Смачивают дистиллированной водой до однородной массы |
|
Чай, кофе |
2,0 |
НNО3 - 8 Н2О2 - 1 |
20 - 30 мин |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
Смачивают дистиллированной водой до однородной массы |
|
Яичный порошок |
2,0 |
НNО3 - 6 Н2О2 - 1 |
30 - 40 мин |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 1 ч |
Смачивают дистиллированной водой до получения однородной массы |
|
8. Молочные продукты |
|||||
|
Сливки |
2,0 |
НNО3 - 6 Н2О2 - 1 |
30 - 40 мин |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
|
|
Кисломолочные продукты (творог, творожные изделия, кефир, йогурт) |
3,0 |
НNО3 - 6 Н2О2 - 1 |
30 - 40 мин |
160 °С - 1 ч 180 °С - 2 ч 200 °С - 1 ч |
|
|
Сухие молочные продукты |
2,0 |
НNО3 - 6 Н2О2 - 1 |
Заливают на ночь НNО3, утром добавляют Н2О2 |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
Смачивают дистиллированной водой до однородной массы |
|
Творог, творожные изделия |
2,5 |
НNО3 - 6 Н2О2 - 1 |
20 - 30 мин |
160 °С - 1 ч 180 °С - 2 ч 200 °С - 1 ч |
|
|
Сыры |
2,5 |
НNО3 - 7 Н2О2 - 1 |
Заливают на ночь НNО3, утром добавляют Н2О2 |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
|
|
Молоко сгущенное |
3,0 |
НNО3 - 6 Н2О2 - 1 |
20 - 30 мин |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
|
|
Мороженое |
5,0 |
НNО3 - 6 Н2О2 - 1 |
- |
160 °С - 1 ч 180 °С - 2 ч |
|
|
9. Растительные масла и животные жиры |
|||||
|
Масла растительные |
1,0 |
НNО3 - 6 Н2О2 - 1 |
Заливают на ночь НNО3, утром добавляют Н2О2 |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
|
|
Масло сливочное |
1,0 |
НNО3 - 6 Н2О2 - 1 |
Заливают на ночь НNО3, утром добавляют Н2О2 |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
|
|
Продукты переработки растительных масел (маргарин, кулинарный жир, майонез) |
2,0 |
НNО3 - 6 Н2О2 - 1 |
Заливают на ночь НNО3, утром добавляют Н2О2 |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 2 ч |
|
|
10. Напитки |
|||||
|
Напитки безалкогольные, соки |
10,0 |
НNО3 - 6 Н2О2 - 1 |
20 мин |
160 °С - 1 ч 180 °С - 2 ч |
|
|
Напитки алкогольные, пиво |
5,0 |
НNО3 - 6 Н2О2 - 1 |
40 - 60 мин |
160 °С - 1 ч 180 °С - 1 ч 200 °С - 1 ч |
Добавлять кислоту небольшими порциями во избежание бурного протекания реакции |
|
Ликеры |
3,0 |
НNО3 - 6 Н2О2 - 1 |
|||
9.3.4.3. При подготовке аналитической пробы не допускается использование инструментов и оборудования, загрязняющих пробу определяемыми элементами.
9.3.4.4. Навеску пробы помещают в реакционную емкость, добавляют смесь реактивов и выдерживают при комнатной температуре согласно табл. 9.1.
9.3.4.5. Для приготовления раствора контрольного («холостого») опыта в реакционную емкость помещают только смесь реактивов без добавления испытуемой пробы.
9.3.4.6. Реакционную емкость закрывают крышкой и герметизируют в металлическом корпусе автоклава.
9.3.4.7. Автоклавы помещают в холодные термостаты, устанавливают на пульте управления температуру, согласно табл. 9.1, и нагревают автоклавы в течение времени, указанного в табл. 9.1
Время выдерживания при минимальной температуре (160 °С) отсчитывается с момента достижения этой температуры в печи, что фиксируется установлением стрелки датчика на нулевую отметку.
9.3.4.8. По окончании минерализации автоклавы извлекают из термостата с помощью приспособления для переноса, помещают в устройство для охлаждения и охлаждают до комнатной температуры (30 - 60 мин, в зависимости от режима нагрева и состава продукта), после чего проводят разгерметизацию автоклава.
9.3.4.9. Полученный минерализат количественно переносят из реакционной камеры автоклава в мерную колбу, вместимостью 25 см3, и доводят до метки дистиллированной водой. Раствор минерализата должен быть бесцветным. Наличие желтой окраски свидетельствует о неполной минерализации.
9.3.4.10. В случае получения желтого раствора после автоклавного разложения (неполная минерализация) минерализат количественно переносят в фарфоровый или стеклянный тигель (чашку), ополаскивая автоклав и крышку небольшим количеством бидистилированной (деионизованной) воды, и осторожно упаривают на водяной бане досуха.
9.3.4.11. Полученный после упаривания остаток коричневого или черного цвета осторожно обрабатывают на водяной бане небольшим количеством (0,5 - 1,0 см3) раствора азотной кислоты (1:1) и несколькими каплями пероксида водорода и снова упаривают до сухих солей. Обработку повторяют до тех пор, пока зола не станет белой или слегка окрашенной, без обугленных частиц.
9.3.4.12. Переведение золы в раствор проводят в соответствии с п. 9.3.1.7.
9.4. Разбавление растворов минерализата
9.4.1. Разбавление растворов минерализата пробы производят в соответствии с массой навески пробы (М, г) и фактором разбавления (k), максимальные значения которого для прибора «Квант-Z.ЭТА» приведены в табл. 9.2. Фактор разбавления выбирается в зависимости от содержания определяемого элемента в пробе и чувствительности прибора. Необходимо помнить, что содержание определяемого элемента в полученном после разбавления анализируемом растворе должно находиться в пределах градуировки прибора.
9.4.2. Перестраиваемой микропипеткой (200 - 1000 мкл) отбирают аликвоту (1000 - 1000 eМ/k) мкл бидистиллированной воды и переносят ее в пробирку типа «Эппендорф», вместимостью 1,5 - 2,0 см3, затем перестраиваемой микропипеткой (10 - 100 мкл) отбирают аликвоту 1000 M/k мкл раствора минерализата пробы (п. 9.3.4) и также помещают ее в пробирку. Полученный раствор тщательно перемешивают. Перестраиваемой микропипеткой (1 - 10 мкл) отбирают аликвоту 2 мкл раствора нитрата палладия с массовой концентрацией 10 мг/см3 (п. 8.2.2), переносят в пробирку с разбавленным раствором минерализата и тщательно перемешивают. Полученный раствор анализируют.
9.4.3. Перестраиваемой микропипеткой (200 - 1000 мкл) отбирают аликвоту 1000 мкл полученного по п. 9.3.4 раствора контрольного опыта и переносят ее в пробирку типа «Эппендорф». Перестраиваемой микропипеткой (1 - 10 мкл) отбирают аликвоту 2 мкл раствора нитрата палладия с массовой концентрацией 10 мг/см3 (п. 8.2.2), переносят в пробирку с раствором холостого опыта и тщательно перемешивают. Полученный раствор анализируют.
Рекомендуемые факторы разбавления (k) раствора минерализата для различных групп пищевых продуктов
|
Элемент |
||
|
Рb |
Сd |
|
|
1 |
2 |
3 |
|
Мясо и птица |
10 М |
10 М |
|
Колбасы и кулинарные изделия из мяса и птицы |
10 М |
10 М |
|
Консервы мясные и мясорастительные |
10 М |
10 М |
|
Субпродукты животных и птиц |
12 М |
60 М |
|
Почки и продукты их переработки |
20 М |
200 М |
|
Яйца |
6 М |
2 М |
|
Яичный порошок |
60 М |
20 М |
|
Молоко и кисломолочные изделия |
2 М |
6 М |
|
Молоко сгущенное |
6 М |
6 М |
|
Молоко сухое |
2 М |
6 М |
|
Сыры и творожные изделия |
6 М |
10 М |
|
Рыба свежая, охлажденная, мороженая, консервы |
20 М |
40 М |
|
Икра, моллюски и ракообразные |
200 М |
400 М |
|
Хлеб |
6 М |
10 М |
|
Зерновые и зернобобовые, мука, крупы, макаронные изделия, бараночные и сухарные изделия |
10 М |
20 М |
|
Сахар-песок, какао, шоколад |
20 М |
10 М |
|
Конфеты и подобные изделия |
20 М |
20 М |
|
Кофе |
20 М |
10 М |
|
Печенье |
10 М |
20 М |
|
Овощи, фрукты, ягоды свежие, свежемороженые и консервированные, грибы |
10 М |
6 М |
|
Чай |
200 М |
200 М |
|
Специи и пряности |
100 М |
40 М |
|
Масло растительное, сливочное, маргарин, животные жиры |
2 М |
10 М |
|
Соль поваренная |
40 М |
20 М |
|
Крахмал |
10 М |
20 М |
|
Желатин |
40 М |
6 М |
9.5. Измерение концентрации элементов
9.5.1. Измерение концентрации элемента в разбавленном испытуемом растворе минерализата пробы и в растворе контрольного («холостого») опыта производят согласно техническому описанию и инструкции по эксплуатации АА спектрометра.
9.5.2. Аликвоту анализируемой пробы объемом 5 мкл (при использовании прибора «Квант-Z.ЭТА») с помощью микропипетки вводят в графитовую печь электротермического атомизатора и включают программу нагрева графитовой печи. При использовании других спектрометров выбор аликвоты производят в соответствии с рекомендациями Руководства по эксплуатации.
9.5.3. После окончания программы нагрева и индикации результатов измерения на дисплее персонального компьютера считывают значение массовой концентрации элемента в анализируемом растворе.
9.5.4. Измерение концентрации в данной пробе производят два раза, регистрируя измеренные значения Х1 и Х2. За результат измерения принимают среднее арифметическое значение:
Расхождение между значениями Х1 и Х2 не должно превышать 5 %, т.е.:
![]() . (3)
. (3)
Если условие (3) не выполняется, измерение повторяют. При повторном нарушении условия (3) выясняют причины, приводящие к неудовлетворительным результатам, и устраняют их.
10. Вычисление результата анализа
Массовую долю элемента в пробах пищевых продуктов и пищевого сырья в мг/кг рассчитывают по формуле:
![]() , где (4)
, где (4)
Х - массовая концентрация элемента в анализируемом растворе, мкг/дм3;
Х0 - массовая концентрация элемента в растворе контрольного опыта, мкг/дм3;
k - фактор разбавления, использованный при подготовке раствора пробы к анализу;
V - объем раствора минерализата, в который переведена навеска, дм3;
М - навеска пробы, г.
За результат анализа принимают среднее арифметическое значение результатов двух параллельных определений m1 и т2.
![]() (5)
(5)
если расхождение между ними не превышает норматива оперативного контроля сходимости d:
![]() , где (6)
, где (6)
![]() . Значения
. Значения ![]() приведены в табл. 2.3.
приведены в табл. 2.3.
При превышении норматива оперативного контроля сходимости измерения повторяют. При повторном превышении норматива d выясняют причины, приводящие к неудовлетворительным результатам, и устраняют их.
11. Форма представления результата анализа
Результат измерения массовой доли элемента в пробах пищевых продуктов и пищевого сырья в документах, предусматривающих его использование, представляют в виде:
![]() мг/кг, где (7)
мг/кг, где (7)
![]() - массовая доля элемента в
продукте, мг/кг;
- массовая доля элемента в
продукте, мг/кг;
D - абсолютная погрешность измерений массовой
доли элемента (![]() ), мг/кг, при доверительной вероятности Р = 0,95.
), мг/кг, при доверительной вероятности Р = 0,95.
Значение D рассчитывают по формуле:
![]() , где (8)
, где (8)
![]() - значение относительной
погрешности измерения массовой доли элемента, приведенное в табл. 2.3.
- значение относительной
погрешности измерения массовой доли элемента, приведенное в табл. 2.3.
Значения массовой доли
элемента в пробе (![]() ) и абсолютной погрешности (D) должны содержать
одинаковое число знаков после запятой.
) и абсолютной погрешности (D) должны содержать
одинаковое число знаков после запятой.
12. Контроль погрешности измерений
Контроль точности результатов анализа проводят согласно МИ 2335 [3].
12.1. Оперативный контроль сходимости
Оперативный контроль сходимости проводят для каждой анализируемой пробы пищевого продукта в соответствии с алгоритмом, приведенным в п. 10.
12.2. Оперативный контроль воспроизводимости
Образцами для контроля
являются реальные пробы пищевых продуктов и пищевого сырья. Масса отобранной
для контроля пробы должна соответствовать удвоенной массе, необходимой для
проведения анализа по методике. Отобранную массу делят на две равные части и
анализируют в точном соответствии с прописью методики в разное время, получая
два результата (первичный - ![]() 1, и повторный -
1, и повторный - ![]() 2),
причем в этом случае максимально варьируют условия проведения анализа; если
возможно - анализ выполняют разные операторы на разных приборах.
2),
причем в этом случае максимально варьируют условия проведения анализа; если
возможно - анализ выполняют разные операторы на разных приборах.
Оперативный контроль
воспроизводимости проводят путем сравнения результата контрольной процедуры Dk, равного
расхождению между двумя полученными результатами измерений (![]() 1 и
1 и ![]() 2) с нормативом оперативного контроля
воспроизводимости D, т.е.:
2) с нормативом оперативного контроля
воспроизводимости D, т.е.:
![]() , где (9)
, где (9)
![]() . Значения
. Значения ![]() приведены в
табл. 2.3.
приведены в
табл. 2.3.
При превышении норматива оперативного контроля воспроизводимости эксперимент повторяют. При повторном превышении норматива D выясняют причины, приводящие к неудовлетворительным результатам контроля, и устраняют их.
12.3. Оперативный контроль погрешности (точности) методики
Оперативный контроль погрешности методики осуществляют с использованием образцов для контроля или методом добавок известного количества определяемого элемента в реальные пробы пищевых продуктов.
12.3.1. Алгоритм проведения оперативного контроля погрешности (точности) с использованием образцов для контроля
Образцами для оперативного контроля точности являются стандартные образцы состава пищевых продуктов или аттестованные смеси, приготовленные в соответствии с МИ 2334 [4]. Погрешность аттестованного значения определяемого элемента в образце для контроля не должна превышать третьей части величины погрешности методики для этого значения.
Алгоритм проведения
оперативного контроля точности с применением образцов для контроля состоит в
сравнении результата контрольной процедуры Кк, равного
разности между результатом контрольного измерения аттестованной характеристики
в образце для контроля ![]() и его
аттестованным значением тко с нормативом оперативного
контроля точности - К.
и его
аттестованным значением тко с нормативом оперативного
контроля точности - К.
Точность результата
контрольного измерения (![]() ), а также точность результатов анализа рабочих проб,
выполненных за период, в течение которого условия проведения анализа принимают
стабильными и соответствующими условиям проведения контрольного измерения,
признают удовлетворительной, если:
), а также точность результатов анализа рабочих проб,
выполненных за период, в течение которого условия проведения анализа принимают
стабильными и соответствующими условиям проведения контрольного измерения,
признают удовлетворительной, если:
![]() , где (10)
, где (10)
К - норматив оперативного контроля погрешности.
Норматив оперативного контроля погрешности рассчитывают по формулам:
· при проведении внутрилабораторного контроля (Р = 0,90):
· при проведении внешнего контроля (Р = 0,95):
При превышении норматива оперативного контроля погрешности контрольный эксперимент повторяют. При повторном превышении указанного норматива погрешности выясняют причины, приводящие к неудовлетворительным результатам контроля, и устраняют их.
12.3.2. Алгоритм проведения оперативного контроля погрешности (точности) с использованием метода добавок
Масса отобранной для контроля рабочей пробы должна соответствовать удвоенной массе, необходимой для проведения анализа по методике. Отобранную массу пробы делят на две равные части, первую из которых анализируют в точном соответствии с прописью методики и получают средний из 2-х параллельных определений результат анализа исходной рабочей пробы - m; ко второй делают добавку определяемого элемента (с использованием аттестованных градуировочных растворов), причем величина добавки должна составлять 50 - 150 % от содержания элемента в исходной пробе. Пробу с добавкой анализируют в точном соответствии с прописью методики, получая средний из 2-х параллельных определений результат анализа рабочей пробы с добавкой - тg Результат анализа пробы с введенной добавкой не должен выходить за верхнюю границу определяемых содержаний.
Сравнивают результат
контрольной процедуры (Кк), равный разности между результатом
контрольного измерения пробы с добавкой (тg),
пробы без добавки (![]() ) и величиной добавки (g) с нормативом оперативного
контроля погрешности (Кд).
) и величиной добавки (g) с нормативом оперативного
контроля погрешности (Кд).
Результаты оперативного контроля погрешности методики считают удовлетворительными, если выполняется условие:
![]() . (13)
. (13)
Норматив оперативного контроля погрешности (Кд) рассчитывают по формулам:
· при проведении внутрилабораторного контроля (Р = 0,90):
![]() , где (14)
, где (14)
D1 и D2 - абсолютные погрешности определения массовой доли элемента в пробе без добавки и с добавкой соответственно, рассчитываемые по формуле (8), для значений т и тg
· при проведении внешнего контроля (Р = 0,95):
![]() . (15)
. (15)
В случае если значение массовой доли элемента в исходной пробе продукта меньше нижней границы диапазона измерений, то добавка должна составлять величину, в 2 - 5 раз превышающую концентрацию, соответствующую нижней границе диапазона измерений элемента. В этом случае норматив оперативного контроля погрешности методики (для Р = 0,90) рассчитывают по формуле:
D - абсолютная погрешность определения массовой доли элемента в пробе с добавкой.
При превышении норматива оперативного контроля погрешности измерения повторяют. При повторном превышении норматива Кд выясняют причины, приводящие к неудовлетворительным результатам контроля, и устраняют их.
12.4. Периодичность контроля устанавливается самой лабораторией с учетом фактического состояния аналитических работ.
13. Список литературы
1. ГОСТ 26929-94. Сырье и продукты пищевые. Подготовка проб. Минерализация для определения токсичных элементов.
2. МУК 4.1.985-00. Определение содержания токсичных элементов в пищевых продуктах и продовольственном сырье. Методика автоклавной пробоподготовки.
3. МИ 2335-95. Рекомендация. ГСИ. Внутренний контроль качества результатов количественного химического анализа.
4. МИ 2334-95. Рекомендация. ГСИ. Смеси аттестованные. Общие требования к разработке.