 | Информационная система | |
МИНИСТЕРСТВО
ЭНЕРГЕТИКИ И ЭЛЕКТРИФИКАЦИИ СССР
ГЛАВНОЕ ТЕХНИЧЕСКОЕ УПРАВЛЕНИЕ
ПО ЭКСПЛУАТАЦИИ ЭНЕРГОСИСТЕМ
МЕТОДИЧЕСКИЕ УКАЗАНИЯ
ПО ХИМИЧЕСКОМУ АНАЛИЗУ ОТЛОЖЕНИЙ
С ВНУТРЕННИХ ПОВЕРХНОСТЕЙ НАГРЕВА
И ПРОТОЧНОЙ ЧАСТИ ТУРБИН
МУ 34-70-102-85
![]()
СОЮЗТЕХЭНЕРГО
Москва 1985
РАЗРАБОТАНО предприятием «Южтехэнерго» Производственного объединения по наладке, совершенствованию технологии и эксплуатации электростанций и сетей «Союзтехэнерго»
ИСПОЛНИТЕЛИ Р.Н. АКСЕЛЬРУД, Г.П. ГОЛЫШЕВА, Л.Г. МАДОЯН
УТВЕРЖДЕНО Главным техническим управлением по эксплуатации энергосистем 16.05.85 г.
Заместитель начальника Д.Я. ШАМАРАКОВ
|
МУ 34-70-102-85 |
Срок действия установлен
с 01.01.86 г.
до 30.11.95 г.
В настоящих Методических указаниях приведены методы определения массовой дожи основных компонентов в отложениях, образующихся на внутренних поверхностях теплосилового оборудования.
Выявлена возможность ускорить и упростить определение массовой доли железа, меди, цинка, никеля, алюминия, кальция, магния, марганца, фосфатов и сульфатов, выполняя анализы раствора отложений преимущественно без отделения определяемого вещества от сопутствующих мешающих компонентов. Создается среда, в которой исключается их влияние, или они связываются в соединения, в присутствии которых возможно выполнение данных анализов.
Использованы методы анализов ВТИ, Южтехэнерго, Мосэнерго и другие материалы.
1. ПОДГОТОВКА ПРОБЫ
Пробу отложений первоначально грубо измельчают, квартованием сокращают до нескольких граммов и растирают в агатовой или яшмовой ступке. Котельные отложения измельчают тщательно до мелкодисперсного состояния («пудры»).
Если при осмотре и измельчении пробы обнаруживаются частички сварочного грата, металлической стружки или иные загрязнения, их удаляют вручную. При этом нельзя пользоваться магнитом, так как окислы железа, содержащиеся в отложениях, также извлекаются магнитом.
2. ОПРЕДЕЛЕНИЕ ПОТЕРИ ИЛИ УВЕЛИЧЕНИЯ МАССЫ ПРИ ПРОКАЛИВАНИИ
В результате прокаливания масса накипи может уменьшиться или увеличиться. Уменьшение массы происходит вследствие потери массовой доли влаги, сгорания органических веществ, разложения карбонатов и т.д. Увеличение массы вызывается окислением кислородом воздуха закиси железа и металлической меди до их окислов.
Точную массу навески (0,5 - 1 г) отложений помещают в прокаленный и взвешенный фарфоровый тигель и прокаливают в муфельной печи при 800 - 850 °С до постоянной массы.
Прокаливание в никелевом, серебряном или платиновом тиглях недопустимо: никелевый тигель при этом сильно окисляется и загрязняет массу навески окисью никеля, серебряный может расплавиться, а платиновый будет испорчен, если в анализируемом материале относительно много металлической меди.
Если при этой температуре отложения плавятся, то берут новую массу навески и прокаливают при температуре 600 °С.
Тигель с массой навески устанавливают в холодную муфельную печь, постепенно повышая температуру до указанной, после чего прокаливают в течение 2 - 3 ч. Тигель с прокаленной массой навески охлаждают в эксикаторе, взвешивают и прокаливают вторично в течение 1 ч. Если масса изменится более чем на 0,5 мг, пробу прокаливают дополнительно.
Потерю при прокаливании в (ПП) вычисляют по формуле, %
где В1 - масса тигля с навеской до прокаливания, г;
В2 - масса тигля с навеской после прокаливания, г;
В - масса навески накипи, г.
Если имеется привес, его вычисляют, подставляя в формулу (1) разность (В2 - В1).
3. РАЗЛОЖЕНИЕ МАССЫ НАВЕСКИ ОТЛОЖЕНИЙ
В зависимости от состава отложений перевод отобранной пробы в растворимое состояние производится обработкой ее водой, соляной кислотой, царской водкой или сплавлением со щелочью.
Отложения, образующиеся в трубках пароперегревателей и проточной части турбин, в основном состоят из водорастворимых соединений натрия, а также содержат нерастворимые в воде силикаты и продукты коррозии.
Отложения, образующиеся при нагревании воды в трубках конденсаторов турбин и различных подогревателях, так называемые низкотемпературные накипи, обычно нерастворимы в воде, но разлагаются соляной кислотой или царской водкой.
Отложения, образующиеся на внутренних поверхностях нагрева котлов, значительная часть которых нерастворима в кислотах, для перевода в растворимое состояние подвергаются сплавлению со щелочью.
Для определения массовой доли водорастворимых веществ массу навески воздушно-сухих измельченных отложений (0,5 - 1 г) помещают в химический стакан, вливают в него 50 см3 горячей дистиллированной воды, образовавшиеся комочки раздавливают и растирают стеклянной палочкой и кипятят жидкость на слабом огне в течение 10 - 15 мин, периодически помешивая ее.
Содержимое стакана количественно переводят в мерную колбу вместимостью 500 см3, смывая туда также оставшиеся частички отложений со стенок стакана струей горячей дистиллированной воды. Жидкости в закрытой колбе дают остыть, после чего доливают до метки дистиллированной водой, содержимое колбы периодически тщательно перемешивают и оставляют отстаиваться. Из хорошо осветлившейся жидкости, не перемешивая ее, отбирают пробы для определения щелочности, жесткости, кальция, магния, сульфатов, хлоридов, силикатов, фосфатов по методам, изложенным в [1 - 4], и натрия пламяфотометрически [1, 2] в соответствии с инструкцией, прилагаемой к прибору.
Если нет полного осветления жидкости, то анализы выполняют из отфильтрованной пробы.
Для анализа нерастворимой в воде части отложений используется осадок, полученный после отделения водорастворимых веществ фильтрованием через беззольный фильтр, прокаливанием и последующим растворением осадка в соляной кислоте, царской водке или сплавлением со щелочью.
Если анализируются отложения с высоким содержанием не растворимых в воде веществ, то отложения для перевода в растворимое состояние обрабатывают кислотой или подвергают предварительному сплавлению со щелочью.
Массу навески (0,5 - 1 г) воздушно-сухих отложений или полученных после определения массовой доли потери при прокаливании количественно переносят в химический стакан, смывая 30 - 50 см3 разбавленной (1:1) соляной кислотой. Жидкость кипятят около 10 мин, не допуская бурного течения реакции во избежание разбрызгивания раствора. Если отложения полностью не растворятся в соляной кислоте, то добавляют 0,33 объема концентрированной азотной кислоты, создавая таким образом раствор царской водки. В случае неполного растворения отложений в царской водке производят сплавление со щелочью отфильтрованного нерастворенного остатка или сплавляют со щелочью всю массу навески отложений.
Разложение отложений для полного перевода их в растворимое состояние может быть выполнено сплавлением с едкими натром или калием, содой, или смесью соды с поташом в соотношении 1:1. Сплавление с едким натром более эффективно, оно выполняется при 500 - 600 °С в течение 30 - 40 мин; сплавление с содой нужно выполнять в платиновом тигле [1 - 3, 5] при температуре 900 - 950 °С в течение 2 - 3 ч. Поэтому предпочтительно производить разложение отложения сплавлением с едким натром.
Сплавление с едким натром выполняют в никелевых или серебряных тиглях. Применять для этого платиновые тигли нельзя, так как при сплавлении с едким натром происходит заметное растворение платины, что приводит к ее потере и затрудняет дальнейшее выполнение анализа.
Никелевый или серебряный тигель, если он еще не был в употреблении, на одну треть заполняют твердым едким натром и ставят на 30 - 40 мин в муфельную печь, нагретую до 400 - 600 °С. После этого тигель извлекают, дают остыть и выщелачивают получившийся сплав дистиллированной водой.
В чистый тигель берут массу навески сухого едкого натра (квалификации «х.ч.») из расчета 3 - 4 г на 1 г массы навески отложений, помещают в холодную муфельную печь и нагревают до 400 - 600 °С. Когда щелочь расплавится (обычно за 2 - 3 мин), тигель вынимают из муфельной печи. В остывший тигель на затвердевшую щелочь переносят массу навески прокаленной накипи из фарфорового тигля. Количество перенесенной накипи проверяют взвешиванием фарфорового тигля. Если требуется ускорить выполнение анализа, производят сплавление со щелочью воздушно-сухой пробы отложений, а потерю при прокаливании определяют из отдельной массы навески.
Тигель со щелочью и массой навески отложений помещают в муфельную печь, повышают температуру до 500 - 600 °С и выдерживают 30 - 40 мин. Затем тигель из печи осторожно вынимают. Если сплав однородный, то сплавление считают оконченным. В противном случае выдерживают тигель в муфеле еще 10 - 20 мин (обычно этого не требуется, если накипь была хорошо измельчена).
Извлечение сплава производят следующим путем. Соляная кислота растворяет металлический никель. Концентрированный раствор щелочи, особенно горячий, воздействует на фарфор и стекло. Поэтому необходимо избегать воздействия кислоты на никель и, по возможности, сокращать продолжительность действия щелочи на фарфор и стекло. Учитывая это, извлечение сплава из никелевого тигля рекомендуется производить водой. В еще теплый, но не горячий тигель с массой навески наливают дистиллированную воду примерно до половины тигля и осторожно подогревают, помешивая содержимое стеклянной палочкой. Жидкость из тигля выливают по папочке в фарфоровую чашку, в которую предварительно наливают 20 - 30 см3 соляной кислоты, разбавленной 1:1. Если сплав полностью не растворился, эту операцию повторяют до полного растворения и перенесения сплава в фарфоровую чашку. Стеклянную палочку не следует оставлять в тигле, а надо, помешав его содержимое, вынимать ее и класть в фарфоровую чашку, куда переносится растворяющийся сплав. Сливать жидкость из тигля в чашку надо быстро, не допуская выливания жидкости на наружную поверхность тигля. Если это произошло, надо смыть эти капли небольшим количеством дистиллированной воды в фарфоровую чашку. Перед началом выщелачивания сплава наружную поверхность тигля нужно промыть дистиллированной водой. При сплавлении пробы отложений с едким натром и вымывании сплава водой (без добавления кислоты) из никелевого тигля переходит в раствор незначительное количество никеля, не мешающее выполнению нижеприведенных анализов.
Соляная кислота почти не действует на серебро. Серебряный тигель, обмыв его наружную поверхность, можно опускать в разбавленную соляную кислоту и кипятить в ней до полного растворения сплава.
Если разложение отложений производится водой или соляной кислотой, то при определении содержания меди и железа для их окисления в анализируемые пробы исходного раствора следует добавлять несколько кристаллов персульфата аммония, несколько капель пергидроля или азотной кислоты.
4. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ КРЕМНЕКИСЛОТЫ
Раствор отложения, полученный обработкой массы навески соляной кислотой или царской водкой или после сплавления пробы со щелочью, выпаривают в фарфоровой чашке на кипящей водяной бане досуха. Затем в чашку наливают 10 - 15 см3 концентрированной соляной кислоты и выпаривают до получения сухого остатка. Такую обработку соляной кислотой для перевода кремниевой кислоты в нерастворимое состояние выполняют 2 - 3 раза. После этого в охлажденную чашку вливают 5 - 10 см3 концентрированной соляной кислоты и 15 - 20 см3 горячей дистиллированной воды, нагревают на водяной бане, перемешивают, добиваясь полного растворения солей, и через несколько минут фильтруют через плотный беззольный фильтр «синяя лента». Для ускорения фильтрования на фильтр помещают небольшое количество фильтробумажной (мацерированной) массы [6]. Фильтрат собирают в мерную колбу вместимостью 500 см3. Фильтрат должен быть совершенно прозрачным. Стенки чашки и осадок на фильтре промывают кипящей дистиллированной водой, подкисленной соляной кислотой (2 - 3 см3 концентрированной соляной кислоты на 250 см3), до отрицательной реакции на трехвалентный ион железа (проба с 10 %-ным раствором роданистого аммония) и затем горячей дистиллированной водой до отрицательной реакции на ион хлора (проба с 1 %-ным раствором нитрата серебра, подкисленным азотной кислотой). Чашку вытирают кусочками беззольного фильтра, которые переносят на фильтр. Промытый осадок вместе с фильтром слегка подсушивают в воронке в сушильном шкафу и переносят в прокаленный и взвешенный фарфоровый тигель, доведенный до постоянной массы, высушивают, озоляют и прокаливают в течение 1,5 - 2 ч при температуре 950 - 1000 °С с повторным прокаливанием в течение 30 - 40 мин до постоянной массы.
Содержание кремнекислоты в процентах в отложении вычисляют по формуле
где В2 - масса тигля с двуокисью кремния, г;
В1 - масса пустого тигля, г;
В - масса навески накипи, г.
Осадок кремнекислоты бывает загрязнен следами железа, алюминия и другими примесями. Если анализ необходимо выполнить точно, то определяют содержание кремнекислоты по потере массы, удалив кремний в виде летучего тетрафторида SiF4, что выполняют только в платиновом тигле. Для этого после взвешивания прокаленный осадок двуокиси кремния смачивают в тигле несколькими каплями дистиллированной воды, вводят около 0,5 см3 концентрированной серной кислоты и, когда тигель остынет, добавляют 5 - 7 см3 чистой концентрированной плавиковой кислоты и содержимое тигля осторожно перемешивают вращением тигля. Раствор в тигле медленно выпаривают сначала на водяной бане, потом на плитке. После прекращения выделения белого дыма (серного ангидрида SO3) тигель с остатком прокаливают при 950 - 1000 °С в течение 30 - 40 мин, охлаждают в эксикаторе и взвешивают. Когда имеются опасения, что не вся двуокись кремния превратилась в тетрафторид, обработку кислотами с выпариванием и прокаливанием повторяют. Остаток в тигле, если масса его значительна, растворяют в соляной кислоте, и раствор присоединяют к фильтрату от кремнекислоты.
Содержание кремнекислоты (SiO2) вычисляют по формуле, %
где B1 - масса тигля с осадком загрязненной двуокиси кремния, г;
В2 - масса тигля после удаления тетрафторида SiF4, г;
В - масса навески накипи, г.
Остаток после удаления кремнекислоты обычно небольшой. Поэтому проверочное удаление кремнекислоты производят редко.
Фильтрат от кремнекислоты, содержащий все катионы, а также анионы фосфорной и серной кислот, которые были в накипи, доводят дистиллированной водой в мерной колбе вместимостью 500 см3 до метки и перемешивают. Полученный раствор именуется исходным или основным раствором накипи. Его используют для определения других компонентов накипи.
5. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ЖЕЛЕЗА
Содержание железа определяется титрованием трилоном Б в присутствии сульфосалицилат-ионов при значении рН раствора 1-2. Присутствие ионов кальция, магния, алюминия, цинка, марганца, никеля, фосфатов, сульфатов не мешает выполнению анализа. Медь не образует с сульфосалицилат-ионами окрашенного соединения, но в присутствии железа при значении рН - 2 почти количественно титруется трилоном. При значении рН = 1 медь значительно меньше завышает результаты определения содержания железа. При значении рН менее 1 разрушение трилоном соединений железа с индикатором идет медленно, и переход окраски индикатора затягивается. При высокой концентрации сильных кислот подавляется диссоциация слабой сульфосалициловой кислоты, и окрашенное в вишневый цвет комплексное соединение трехвалентного железа не образуется.
Влияние меди на определение массовой доли железа существенно уменьшается, если анализируется раствор со значением рН = 1 и с небольшим количеством индикатора сульфосалициловой кислоты.
Так, например, при анализе раствора с рН = 1, введении 0,1 см3 20 %-ной сульфосалициловой кислоты и массовой доле меди в пробе 3 - 10 мг завышаются результаты определения железа соответственно примерно на 0,2 - 0,6 мг, что при массе навески отложения 1 г, массовой доле меди 5 - 50 %, объеме исходного раствора 500 см3 и объеме пробы на анализ 10 см3 завышаются данные определения массовой доли железа в отложениях на 1 - 3 %.
Приведенные данные указывают, что обычно можно определять массовую долю железа, не отделяя его от меди. Если же массовая доля меди относительно велика и анализ требуется выполнить по возможности более точно, то определение железа производится после осаждения его аммиаком, отделения фильтрованием и растворения соляной кислотой. Обычно для полного отделения железа от меди производится двукратное осаждение и отделение полуторных окислов добавлением аммиака в небольшом избытке [1-3]. При этом осадок полуторных окислов может быть использован для трилонометрического определения массовой доли железа и затем алюминия [1], а фильтрат - для определения суммарной массовой доли меди и цинка, а затем суммарной массовой доли кальция и магния [3].
Отделение железа от меди аммиаком можно значительно ускорить однократным осаждением железа, если добавлять аммиак в большом избытке и операции осаждения и фильтрования объединить в одну [7] как описано ниже.
Определение массовой доли железа после его отделения от меди лучше производить при значении рН ≈ 2, так как при этом переход окраски индикатора более четкий, чем при рН = 1.
Ввиду малой скорости взаимодействия железа с трилоном Б раствор нагревают перед титрованием до 60 - 70 °С.
Реактивы:
Трилон Б, 0,05-молярный (0,1 - нормальный) или 0,005 - молярный (0,01 - нормальный) растворы.
Сульфосалициловая кислота, 20 %-ный раствор.
Соляная кислота, раствор 1:1.
Аммиак, 23 %-ный раствор.
Аммиак, растворы 1:1 и 1:20.
Выполнение определения:
из исходного раствора отложений отбирают в коническую колбу в зависимости от предполагаемой массовой доли железа 10 - 25 см3 и добавляют для гарантии полного окисления железа несколько капель концентрированной азотной кислоты, перекиси водорода или несколько кристаллов персульфата аммония. Определить по индикаторной бумаге с достаточной точностью значение рН раствора, равное 1, невозможно, поэтому доведение его до указанного значения выполняют следующим путем. Пробу нагревают до кипения и медленно при перемешивании добавляют разбавленный (1:1) раствор аммиака до начала образования осадка гидроксида железа. Добавляют 10 см3 раствора 1 - нормальной соляной кислоты и, когда осадок гидроксида железа растворится, дистиллированной водой доводят объем до 100 см3. После этого анализируемый раствор нагревают до 60 - 70 °С, добавляют 0,1 см3 20 %-ного раствора сульфосалициловой кислоты и титруют 0,05-молярным или 0,005-молярным раствором трилона Б до изменения вишневой или розовой окраски в желто-зеленую или до обесцвечивания (трилонат железа имеет слабую желто-зеленую окраску, заметную лишь при высокой концентрации железа).
Если оттитрованный раствор при стоянии медленно опять розовеет, что может быть из-за наличия меди, на это не следует обращать внимания.
При значительной массовой доле меди производят осаждение железа аммиаком и отделяют его фильтрованием. Для фильтрования применяют обычную воронку с беззольным фильтром «красная лента» или «белая лента» или лучше воронку цилиндрической формы с крупнопористой пластинкой, на которую помещают слой ваты высотой 5 - 10 мм. Нижнее отверстие воронки закрывают полиэтиленовой или резиновой трубкой с винтовым зажимом и устанавливают в штатив над стаканом. В воронку наливают 5 - 10 см3 концентрированного аммиака и в зависимости от предполагаемой массовой доли железа 10 - 25 см3 исходного раствора отложений и оставляют на 10 мин. Затем открывают затвор, отфильтровывают осадок и промывают 3-4 раза горячим раствором аммиака 1:20. Воронку с промытым осадком помещают над конической колбой, осадок растворяют около 5 см3 горячей соляной кислоты 1:1 и промывают несколько раз горячей дистиллированной водой. Доводят объем пробы дистиллированной водой до 100 см3 и добавляют раствор аммиака 1:1 до значения рН ≈ 2. После этого производят определение содержания железа, как указано выше.
Массовую долю железа в пересчете на оксид железа (Fe2O3) вычисляют по формуле, %
где а - расход раствора трилона Б на титрование, см3;
М - молярность раствора трилона Б;
К - поправочный коэффициент к данной молярности;
Vисх - общий объем исходного раствора отложений, см3;
Vпр - объем исходного раствора отложений, взятый для анализа, см3;
В - масса навески отложений, г;
79,85 - эквивалентная масса Fe2O3 в реакции с трилоном Б, равная половине молекулярной массы.
Определение массовой доли железа из исходного раствора накипи можно выполнить также с помощью автоматического титратора (Т-107 или Т-108). Автоматические приборы для титрования позволяют повысить точность и объективность получаемых результатов, а также сократить время анализа.
6. ОПРЕДЕЛЕНИЕ СУММАРНОЙ МАССОВОЙ ДОЛИ МЕДИ И ЦИНКА В ЦИТРАТНОЙ СРЕДЕ
Метод основан на том, что в среде двухзамещенного аммония лимоннокислого (рН ≈ 5) в присутствии индикатора 1-(2-пиридилазо) - 2-нафтол (ПАН) трилоном Б количественно титруется сумма ионов Cu2+ и Zn2+ [6, 9]. В отличие от меди цинк не образует с ПАН в указанной среде окрашенного соединения, поэтому анализ выполним лишь в присутствии меди (11). Экспериментально установлено, что если массовая доля меди в пробе менее 0,1 мг, могут получаться заниженные результаты определения суммарной массовой доли меди и цинка. Во избежание этого, если неизвестно, что меди достаточно, вводят раствор трилоната меди (или точное количество меди, которое затем учитывают, вычитая из расхода трилона на титрование расход его на введенную медь).
Анализ выполним в присутствии железа, алюминия, кальция, магния, никеля. Железо связывается в нитратный комплекс и три лоном Б не титруется. При массовой доле железа в титруемом растворе более 10 мг переход окраски отмечается менее четко, так как в указанной среде наличие железа придает раствору желтую окраску. Ионы Mn2+ замедляют переход окраски индикатора. В их присутствии во избежание получения завышенных результатов анализа титрование раствором трилона Б нужно производить медленно, особенно вблизи эквивалентной точки. Присутствие кальция, магния, алюминия не мешает выполнению анализа. Присутствие никеля не мешает выполнению анализа, если учитывать следующее. При комнатной температуре в среде двухзамещенного лимоннокислого аммония взаимодействие трилона Б с никелем происходит настолько медленно, что в его присутствии наблюдается четкий переход окраски индикатора при окончании титрования цинка и меди. Окраска оттитрованной пробы, содержащей никель, постепенно изменяется, на что не следует обращать внимания. С повышением температуры реакция взаимодействия трилона Б с ионами никеля ускоряется, поэтому для определения массовой доли суммы ионов Cu2+ и Zn2+ в присутствии никеля температура титруемого раствора не должна превышать 20 - 25 °С.
Метод позволяет определить суммарную массовую долю меди и цинка 1 мг в пробе с относительной ошибкой около 10 %. При наличии в отложениях марганца ошибка анализа увеличивается.
Реактивы:
Трилон Б, 0,005-молярный или 0,05-молярный раствор.
Аммиак, раствор 1:1.
Аммоний лимоннокислый двухзамещенный, 20 %-ный раствор
Смешанный индикатор ПАН - смесь 0,08 г ПАН в 80 см3 этилового спирта и 0,02 г метиленового голубого (метиленового синего) в 20 см3 спирта или 0,1 %-ный спиртовой раствор ПАН.
Раствор трилоната меди. Смешивают точно эквивалентные количества 0,1-нормадьных растворов сульфата меди и трилона Б.
Выполнение определения:
отбирают в коническую колбу 10 - 20 см3 исходного раствора отложения, разбавляют дистиллированной водой до 100 см3 и добавлением аммиака доводят до рН ≈ 3. Значение рН определяют по индикаторной бумаге. Добавляют 10 см3 раствора двухзамещенного аммония лимоннокислого, 1 - 2 см3 трилоната меди, 5 - 7 капель индикатора и медленно при интенсивном перемешивании титруют раствором трилона Б до изменения окраски: из фиолетово-сиреневой в желто-зеленую при применении смешанного индикатора; когда применяют ПАН без метиленового голубого окраска изменяется из фиолетово-красной в желтую.
Медь и цинк имеют близкие по значению атомные массы, поэтому, если не определяют раздельно массовые доли меди и цинка, их общую массовую долю в пересчете на оксиды рассчитывают по формуле, %
где а - расход раствора трилона Б на титрование, см3;
М - молярность раствора трилона Б;
K - поправочный коэффициент к данной молярности;
80
- молекулярная масса (усредненная) ![]() .
.
7. ОПРЕДЕЛЕНИЕ СУММАРНОЙ МАССОВОЙ ДОЛИ МЕДИ И ЦИНКА, А ЗАТЕМ НИКЕЛЯ В СРЕДЕ БИФТОРИДА АММОНИЯ
В среде бифторида аммония (рН ≈ 3,7) в присутствии индикатора ПАН и ионов меди (II) количественно титруются трилоном Б ионы меди и цинка, а затем после нагревания титруется никель [8].
Если меди относительно мало, то во избежание получения заниженных результатов анализа вводят трилонат меди (или точное количество меди, вычитая расход трилона при титровании на введенную медь).
Анализ выполним в присутствии железа, кальция, магния, алюминия, марганца.
Фтористые соли разъедают стекло, поэтому для анализов с их применением следует выделить специальную посуду. Однако ввиду отсутствия полиэтиленовых конических колб для титрования допускается использование стеклянных. С одними и тени же стеклянными колбами можно работать несколько месяцев.
Метод позволяет определить суммарную массовую долю меди и цинка 1 мг в пробе с относительной ошибкой около 5 %.
Реактивы:
Трилон Б, 0,005-молярный раствор.
Аммиак, раствор 1:1.
Бифторид аммония, 20 %-ный раствор (приготавливать и хранить в полиэтиленовой посуде).
Раствор индикатора ПАН (см. п. 6).
Раствор трилоната меди (см. п. 6).
Выполнение определения:
отбирают в коническую колбу 10 - 20 см3 исходного раствора отложения, разбавляют дистиллированной водой до 100 см3 и добавлением аммиака доводят до рН ≈ 3 (определяют рН по индикаторной бумаге). Затем добавляют 10 см3 раствора бифторида аммония (отмеряют небольшим протарированным сосудом из оргстекла или полиэтилена), 1 - 2 см3 трилоната меди, 5 - 7 капель индикатора и титруют трилоном Б до перехода окраски: из фиолетово-сиреневой в желто-зеленую при применении смешанного индикатора ПАН; когда применяют раствор ПАН без метиленового голубого, окраска изменяется из красно-фиолетовой в желтую.
Расход трилона Б на титрование соответствует суммарной массовой доле меди и цинка. Если при стоянии раствора постепенно появляется розовый оттенок вследствие медленного взаимодействия индикатора с никелем, на это не следует обращать внимания.
Для определения массовой доли никеля раствор, полученный после определения суммарной массовой доли меди и цинка, нагревают до 70 - 80 °С и, если при этом раствор окрасится в фиолетово-сиреневый или красно-фиолетовый цвет, его титруют трилоном.
Суммарную массовую долю меди и цинка вычисляют по формуле (5).
Массовую долю никеля в пересчете на оксид никеля (NiO) вычисляют по формуле, %.
где δ - расход трилона Б на титрование подогретой пробы, см3;
74,71 - молекулярная масса NiO.
8. ПОСЛЕДОВАТЕЛЬНОЕ ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ МЕДИ И ЦИНКА
Массовую долю меди определяют иодометрически, а затем в той же пробе комплексометрически определяют массовую долю цинка [9, 10].
Двухвалентная медь в слабокислой среде восстанавливается йодистым калием до одновалентной, выделяя из йодистого калия эквивалентное количество йода. Выделившийся йод титруют тиосульфатом натрия в присутствии крахмала, добавляемого в конце титрования.
Для того, чтобы реакция восстановления меди и выделения йода протекала количественно, йодистый калий вводят в большом избытке. Реакцию нужно проводить в слабокислой среде. С повышением рН раствора реакция восстановления меди замедляется и может не дойти до конца. В сильно кислой среде происходит окисление йодид-ионов кислородом воздуха. Требуемая кислотность создается добавлением бифторида аммония или бифторида калия к исходному раствору отложений, предварительно нейтрализованному до значения рН ≈ 3.
Йодометрическому определению массовой доли меди мешают окислители, в том числе трехвалентное железо, которое обычно присутствует в отложениях. Бифторид связывает железо во фторидный комплекс, не мешающий определению меди.
Наличие алюминия, кальция, магния, марганца, никеля, фосфатов в тех количествах и соотношениях, какие обычно бывают в исходных растворах котельных накипей, не мешает йодофторидному определению массовой доли меди.
Анализ выполняют из исходного раствора, получаемого сплавлением массы навески отложения с едким натром в никелевом или серебряном тигле.
Если сплавление массы навески отложения производится в платиновом тигле с карбонатами натрия и калия, то при введении йодистого калия для определения массовой доли меди наблюдается появление не желтой, а желто-вишневой окраски, более яркой при большей массовой доли меди. При титровании пробы тиосульфатом натрия в присутствии крахмала сперва исчезает синяя окраска, остается более слабая желто-вишневая, которая тоже обесцвечивается при дальнейшем добавлении тиосульфата натрия; в этом случае нет уверенности в правильности определения точки конца титрования.
Метод позволяет определить массовую долю меди в пробе с относительной ошибкой около 7 %.
Для определения массовой доли цинка после определения массовой доли меди рН раствора доводят добавлением аммиака до значения 4, проверяя его для большей точности не по индикаторной бумаге, а по так называемому вечернему индикатору. Реакция взаимодействия двухвалентной меди с йодистым калием обратима и при повышении значения рН сдвигается в сторону образования двухвалентной меди. При рН = 4 небольшая часть восстановленной меди опять переходит в двухвалентную, но наличие ее дает возможность определять массовую долю цинка титрованием трилоном Б с индикатором ПАН, так как в создаваемой среде и при указанном значении рН цинк не дает с ПАН красного окрашенного соединения.
Анализами растворов, не содержащих цинка, и растворов с известной массовой долей цинка установлено, что при первоначальной массовой доле в пробах двухвалентной меди в количестве 3 - 10 мг (когда на титрование йода, выделившегося при добавлении йодистого калия расходуется 0,5 - 1,5 см3 0,1-нормального тиосульфата натрия) при добавлении избытка 0,5 см3 раствора тиосульфата натрия и доведении рН раствора до значения 4 опять появляется двухвалентная медь в количестве 0,13 - 0,19 мг (на ее титрование расходуется 0,4 - 0,6 см3 0,005-молярного раствора трилона Б). Если первоначальное количество меди меньше указанного и в пробу после доведения до рН ≈ 4 добавить 1 - 2 см3 0,005-молярного раствора сернокислой меди, то в анализируемом растворе остается двухвалентная медь в том же количестве (0,13 - 0,19 мг).
Для определения массовой доли цинка и внесения правильной поправки на медь нужно по возможности точно доводить значение рН до 4, так как с повышением его значения больше меди переходит в двухвалентную, что завышает результаты анализа определения цинка, а при меньшем его значении будут получены заниженные результаты. Если переход окраски индикатора нечеткий и нет уверенности, что pH раствора приближается к 4, то массовую долю цинка можно вычислить по разности между суммой меди и цинка формулы (5 и 6) и меди формула (7).
При массовой доле в пробе кальция более 5 мг, магния более 3 мг получаются заниженные результаты определения цинка. Наличие двухвалентного марганца в количестве более 1 мг завышает результаты определения цинка. Наличие в пробе никеля более 0,02 мг ухудшает переход окраски индикатора, остается розовый оттенок.
Метод позволяет определить массовую долю цинка 1 мг в пробе с относительной ошибкой около 20 %.
Определение массовой доли меди
Реактивы:
Аммиак, раствор 1:1.
Бифторид аммония (NH4F HF) или бифторид калия (KF · HF), 20 %-ный раствор (хранится в полиэтиленовой посуде).
Йодистый калий, 40 %-ный раствор.
Титрованный раствор тиосульфата натрия 0,1-нормальной или 0,01-нормальной концентрации.
Крахмал растворимый, 0,5 %-ный свежеприготовленный раствор.
Примечание. Растворы йодистого калия и тиосульфата натрия пригодны для анализа в течение недели при условии их хранения в склянке из темного стекла и при комнатной температуре.
Выполнение определения:
отбирают в коническую колбу 10 - 20 см3 исходного раствора отложения, добавляют дистиллированной воды до 50 см3 и доводят до рН ≈ 3 добавлением аммиака (проверяют рН по индикаторной бумаге). Затем добавляют 5 см3 бифторида (раствор при этом обеспечивается), 10 см3 йодистого калия, перемешивают, закрывают колбу часовым стеклом, ставят в темное место и через минуту титруют выделившийся йод тиосульфатом натрия до соломенно-желтого цвета, прибавляют в конце титрования 1 см3 раствора крахмала и продолжают титрование до исчезновения синей окраски. Затем ставят пробу в темное место еще на 1 - 2 мин, чтобы проверить, завершилась ли реакция восстановления меди; при появлении синей окраски дотитровывают тиосульфатом натрия.
Массовую долю меди в пересчете на оксид меди (CuO) вычисляют по формуле, %:
![]() (7)
(7)
где a - расход раствора тиосульфата натрия на титрование, см3;
N - нормальность раствора тиосульфата натрия;
K - поправочный коэффициент к данной нормальности;
79,54 - эквивалентная масса CuO в данной реакции.
Примечание. Так как иодит калия бывает загрязнен иодатом (IO3-) и водные растворы йодистого калия на свету постепенно выделяют свободный йод, проводят «холостой» опыт и, если требуется, вводят соответствующую поправку.
Определение массовой доли цинка
Реактивы:
Тиосульфат натрия, 0,1-нормальный раствор.
Аммиак, раствор 1:1.
«Вечерний индикатор»: растворяют 0,1 г метилового оранжевого и 0,25 г индигокармина в 100 см3 дистиллированной воды. Окраска индикатора: в щелочной среде - зеленая, при рН = 4 (переходная окраска) - серая, в более кислой - фиолетовая.
Смешанный индикатор (см. п. 2.6),
Трилон Б, 0,005-мопярный и 0,05-молярный раствор.
Сульфат меди, 0,005-молярный раствор.
Выполнение определения:
в раствор, полученный после иодометрического определения содержания меди, добавляют избыток 0,5 см3 0,1-нормального раствора тиосульфата натрия, затем капли «вечернего индикатора» и доводят до значения рН ≈ 4, добавляя аммиак до исчезновения розово-фиолетовой и появления серой или почти бесцветной (но не зеленой) окраски.
Если при определении массовой доли меди на титрование йода, выделившегося при восстановлении меди йодистым калием, пошло 5 см3 или более 0,01-нормального (0,5 см3 0,1-нормального) раствора тиосульфата натрия, то после доведения значения рН ≈ 4 вводят 4 - 5 капель смешанного индикатора ПАН и при интенсивном перемешивании медленно титруют раствором трилона Б до изменения окраски из фиолетово-сиреневой в желто-зеленую.
Если при определении меди пошло менее 5 см3 0,01-нормального (0,5 см3 0,1-нормального) раствора тиосульфата натрия, то после доведения рН ≈ 4 добавляют 1 - 2 см3 0,005-молярного раствора сульфата меди, вводят индикатор ПАН и медленно титруют раствором трилона Б.
Массовую долю цинка в пересчете на оксид цинка (ZnO) вычисляют по формуле, %
![]() (8)
(8)
где a - расход раствора трилона Б на титрование, см3;
n - поправка на двухвалентную медь - 0,5 см3, 0,005-молярного или соответственно 0,05 см3 0,05-молярного раствора;
M - молярность раствора трилона Б;
K - поправочный коэффициент к данной молярности;
81,37 - молекулярная масса ZnO в данной реакции.
9. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ МАРГАНЦА
Метод основан на окислении марганца до марганцовой кислоты в кислой среде персульфатом аммония в присутствии кобальтового катализатора. Образующуюся марганцовую кислоту оттитровывают щавелевокислым натрием. Для устранения присущей ионам кобальта розовой окраски, затрудняющей определение эквивалентной точки, к раствору кобальта добавляют соль меди или никеля.
Наличие хлор-ионов мешает анализу, так как они взаимодействуют с марганцовой кислотой, окисляясь до свободного хлора, и восстанавливают марганец до двухвалентного состояния, что ведет к занижению результатов анализа; их удаляют выпариванием раствора с серной кислотой.
Добавление фосфорной кислоты устраняет окраску раствора иона железа (III) и стабилизирует образующуюся марганцовую кислоту, предотвращая возможность ее разложения при кипячении раствора для разложения избытка введенного окислителя. Во избежание большого остатка персульфата аммония он вводится в анализируемый раствор небольшими порциями до полного развития окраски марганцовой кислоты.
Титрование марганцовой кислоты щавелевокислым натрием следует проводить при температуре 55 - 60 °С, так как в этом интервале температур обеспечивается достаточно быстрое их взаимодействие в эквивалентных количествах.
Метод позволяет определить массовую долю марганца 0,5 мг в пробе с относительной ошибкой около 10 %.
Реактивы:
Серная кислота концентрированная.
Ортофосфорная кислота концентрированная.
Персульфат аммония, 20 %-ный раствор, пригоден для анализа в течение 20 дн. при условии хранения в склянках темного стекла при комнатной температуре.
Кобальтово-никелевый катализатор приготавливают растворением 1 г сернокислого кобальта (CoSO4 · 7H2O) и 3 г сернокислого никеля (NiSO4 · 7H2O) в 100 см3 воды, подкисленной 5 - 10 каплями серной кислоты.
Щавелевокислый натрий, 0,1-нормальный раствор, срок хранения приготовленного раствора не более 20 дн.
Выполнение определения:
в коническую колбу на 250 см3 отбирают 50 - 100 см3 исходного раствора отложений, добавляют 5 см3 серной кислоты и упаривают до объема примерно 10 см3 для полного удаления хлор-ионов. В охлажденный раствор прибавляют 3 - 5 см3 ортофосфорной кислоты, дистиллированную воду до объема 100 см3 и 5 см3 кобальтового катализатора. В интенсивно кипящий раствор вводят раствор персульфата аммония (порциями по 0,5 см3) до стойкого неизменяющегося окрашивания раствора, после чего кипятят еще одну минуту. Раствор быстро охлаждают до 60 °С и при интенсивном перемешивании медленно титруют щавелевокислым натрием до исчезновения характерной малиновой окраски.
Содержание марганца в пересчете на оксид марганца (MnO) вычисляют по формуле, %
где а - расход раствора щавелевокислого натрия на титрование, см3;
N - нормальность раствора щавелевокислого натрия;
K - поправочный коэффициент к данной нормальности;
14,18 эквивалентная масса MnO в данной реакции, равная пятой части молекулярной массы.
10. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ АЛЮМИНИЯ
В исходный раствор отложений вводят трилон Б в избытке для образования трилонатов алюминия, железа и других комплексующихся при этом катионов. Избыток трилона Б оттитровывают раствором соли цинка в присутствии индикатора ПАН или ксиленовового оранжевого. Затем к анализируемому раствору добавляют фтористый натр. Комплекс алюминия с трилоном разрушается, алюминий связывается в более прочное соединение Na3AlF6 (криолит). Освободившееся количество трилона Б, эквивалентное содержание алюминия, оттитровывают раствором соли цинка. Во избежание частичного вытеснения трилона Б из трилоната железа не следует вводить в пробу большого избытка фторида натрия.
Метод позволяет определить массовую долю алюминия 1 мг в пробе с относительной ошибкой около 10 %.
Реактивы:
Соляная кислота, раствор 1:1.
Аммиак, раствор 1:1.
Трилон Б, 0,025-молярный раствор.
Натрий фтористый, насыщенный раствор (4 %).
Ацетатный буферный раствор (рН = 6) : 550 г ацетата натрия CH3COONa · 3H2O растворяют в 1 дм3 дистиллированной воды и добавляют 100 см3 раствора уксусной кислоты (который готовят разбавлением 58 см3 ледяной уксусной кислоты до 1 дм3).
Сернокислый цинк, 0,025-молярный раствор: 7,189 г сульфата цинка ZnSO4 · 7H2O растворяют в дистиллированной воде и доводят объем до 1 дм3.
Поправочный коэффициент к молярности приготовленного раствора цинка устанавливают по титрованному раствору трилона Б в аммиачно-буферной среде (рН ≈ 10) с индикатором хром темно-синим.
Раствор индикатора ПАН или ксиленового оранжевого: 0,1 г индикатора ПАН растворяют в 100 см3 этилового спирта;
0,13 г индикатора ксиленолового оранжевого растворяют в 2 см3 1-нормального раствора едкого натра и разбавляют раствор водой до 100 см3.
Выполнение определения:
отбирают в коническую колбу на 500 см3 20 - 50 см3 исходного раствора отложений, добавляют дистиллированной воды до 150 см3 и доводят значение рН до 1 - 2, добавляя аммиак или соляную кислоту. Вводят 10 - 30 см3 раствора трилона Б и медленно нейтрализуют раствором аммиака до рН ≈ 5 ÷ 6 (проверяя по индикаторной бумаге). Вводят 20 см3 ацетатного буферного раствора, кипятят 2 - 3 мин и охлаждают в проточной воде до комнатной температуры. Добавляют 8 - 10 капель раствора индикатора ПАН или ксиленолового оранжевого и титруют окрашенную в зависимости от индикатора в желтый или оранжевый цвет пробу (избыток трилона Б) раствором сернокислого цинка до изменения цвета в фиолетово-красный. Вводят 40 см3 раствора фтористого натрия, кипятят пробу 5 - 7 мин, охлаждают проточной водой. Добавляют 3 - 5 капель индикатора и титруют раствором сернокислого цинка окрасившуюся в желтый или оранжевый цвет пробу (если содержится алюминий) до перехода в фиолетово-красный.
Массовую долю алюминия в пересчете на оксид алюминия (Al2O3) вычисляют по формуле, %
где а - расход раствора цинка на второе титрование, см3;
M - молярность раствора цинка;
K - поправочный коэффициент к данной нормальности;
50,96 - эквивалентная масса Al2O3 в реакции с триионом Б, равная половине молекулярной массы.
11. ОПРЕДЕЛЕНИЕ СУММАРНОЙ МАССОВОЙ ДОЛИ КАЛЬЦИЯ И МАГНИЯ
Определению массовой доли кальция и магния (жесткости) в исходных растворах отложений титрованием трилоном Б в аммиачной буферной среде (рН = 9 ÷ 10) с хромовыми металлиндикаторами мешает наличие железа, меди, цинка, алюминия, марганца, никеля и др. Образующийся осадок гидроксида железа не титруется трилоном, но мешает отмечать изменение окраски индикатора. Ионы цинка количественно титруются совместно с кальцием и магнием. Медь и никель образуют с хромовыми индикаторами более прочные комплексы, чем кальций и магний, затягивая переход окраски индикатора в эквивалентной точке.
Мешающее влияние указанных веществ устраняется введением диэтилдитиокарбамата натрия, с которым они образуют труднорастворимые соединения: медь и никель в сильнокислой среде и до значения рН ≈ 8 ÷ 9; железо - в сильнокислой среде и до рН ≈ 5 ÷ 6; цинк и марганец - при рН ≈ 1,5 ÷ 6. Растворы диэтилдитиокарбамата натрия имеют слабощелочную реакцию. При его добавлении в пробу кислого раствора отложений значение рН повышается, проходя через интервал оптимальных значений рН ≈ 2 ÷ 3, при котором происходит образование диэтилдитиокарбаматов перечисленных мешающих веществ. Комплекс диэтилдитиокарбамата алюминия непрочный. Если массовая доля алюминия превышает 1 мг (в пробе взятой на анализ), то четкость перехода окраски индикатора ухудшается - остается розовый оттенок. Влияние алюминия устраняется введением триэтаноламина.
Исходные растворы отложений содержат обычно такие количества железа и меди, что даже при стократном и большем разбавлении дистиллированной водой добавление диэтилдитиокарбамата натрия приводит к образованию темных жидкостей, которые невозможно титровать. При этом получаются коллоидные растворы и мелкодисперсные осадки, плохо отделяемые фильтрованием. При введении же диэтилдитиокарбамата натрия в пробы исходных растворов отложений до разбавления дистиллированной водой осадки получаются более крупные, а раствор значительно прозрачнее, что дает возможность определить жесткость. Обусловлено это тем, что повышенная суммарная концентрация ионов в растворе уменьшает или полностью нейтрализует заряд коллоидных частиц и улучшает их коагуляцию. Во избежание получения заниженных результатов вследствие возможного соосаждения кальция дозировка диэтилдитиокарбамата натрия не должна превышать 0,1 г на 1 см3 исходного раствора. Чтобы осадок получился более укрупненным, перед добавлением диэтилдитиокарбамата натрия в пробу добавляют электролит - соляную кислоту. Когда осадка много и он затрудняет наблюдение за изменением окраски раствора, его отделяют фильтрованием через беззольный бумажный фильтр или гигроскопическую вату.
Если анализируется проба с малой жесткостью, то фильтрующий материал следует предварительно промыть, как указано ниже. Даже беззольные фильтры часто содержат взаимодействующие с три лоном Б вещества в количествах, которые могут завысить результаты определения жесткости анализируемой пробы на 0,01 - 0,03 миллиграмм-эквивалента (при объеме анализируемой пробы исходного раствора 10 см3, общем его объеме 500 см3 и массе навески отложения 1 г результат анализа в пересчете на массовую долю кальция будет завышен на 1 - 3 %).
Метод позволяет определить массовую долю кальция и магния 0,01 миллиграмм-эквивалента в пробе с относительной ошибкой 10 - 15 %.
Реактивы:
Трилон Б, 0,05-молярный или 0,005-молярный раствор.
Аммиачный буферный раствор 2 %-ный NH4Cl и по NH3 приготавливается смешиванием 80 см3 25 %-ного раствора аммиака со 100 см3 20 %-ного раствора хлористого аммония и доведением объема жидкости до 1 дм3 дистиллированной водой.
Раствор индикатора кислотного хром темно-синего: для его приготовления растворяют 0,5 г индикатора в 20 см3 аммиачного буферного раствора и доводят объем раствора до 100 см3 этиловым спиртом. По данным ПО «Союзтехэнерго», хром темно-синий можно готовить только на аммиачно-буферном растворе.
Диэтилдитиокарбамат натрия кристаллический.
Соляная кислота, раствор 1:1.
Аммиак, раствор 1:1.
Триэтаноламин, 30 %-ный раствор.
Выполнение определения:
отбирают в коническую колбу 5 - 10 см3 (если требуется, то больший объем) исходного раствора отложений, прибавляют 3 - 5 капель соляной кислоты, затем 0,3 - 0,5 г или соответственно 0,5 - 1 г кристаллического диэтилдитиокарбамата натрия. Вводить диэтилдитиокарбамат натрия лучше по частям в 3 - 4 приема. По мере его введения раствор сначала темнеет, затем осадки становятся крупнее, а раствор прозрачнее. После каждого добавления диэтилдитиокарбамата натрия пробу перемешивают, не производя резкого взбалтывания, и оставляют на 2 - 3 мин для укрупнения осадка. Когда осадок получается таким, что можно четко отличать изменение окраски, доводят объем дистиллированной водой до 100 см3, проверяют рН (по индикаторной бумаге) и, если требуется, Доводят его до значения 7 - 9, добавляя раствор аммиака. Затем добавляют 5 см3 аммиачного буферного раствора, 5 - 7 капель индикатора кислотного хром темно-синего и титруют раствором трилона Б до изменения окраски из красной в сине-сиреневую. Если образовавшийся осадок диэтилдитиокарбаматов мешает четко отметить переход окраски, то после добавления диэтилдитиокарбамата натрия проверяют значение рН подученного раствора (по индикаторной бумаге) и во избежание получения заниженных результатов анализа, что наблюдается главным образом при наличии фосфатов, подкисляют раствор соляной кислотой до рН = 2 ÷ 3. Затем осадок отделяют фильтрованием раствора через беззольный фильтр или гигроскопическую вату. Если анализируют пробу с малой жесткостью, производят предварительно промывку фильтрующего материала следующим путем. Беззольный фильтр или вату помещают в воронку, как обычно для фильтрования, и промывают его 100 - 150 см3 подкисленной до значения рН ≈ 2 ÷ 3 дистиллированной водой, подогретой до 50 - 60 °С.
Фильтр с осадком промывают несколько раз дистиллированной водой, объем фильтрата доводят до 100 см3 и продолжают анализ, как указано вине.
Если переход окраски индикатора нечеткий, остается розовый оттенок, что может быть вызвано повышенной массовой долей алюминия, то анализ определения жесткости повторяет, добавляя в пробу перед вводом аммиачно-буферной смеси 20 см3 30 %-ного раствора триэтаноламина. При применении триэтаноламина следует определить поправку на «холостую» пробу и, если она существенна, учесть.
Когда суммарная массовая доля кальция и магния невелика и определять отдельно кальций и магний нецелесообразно, их общую массовую долю в пересчете на оксид кальция (СаО) вычисляют по формуле, %
где a - расход трилона Б на титрование, см3;
М - молярность раствора трилона Б;
К - поправочный коэффициент к данной молярности;
56,08 - молекулярная масса CaO.
Примечание. При наличии фосфатов титрование трилоном Б нужно производить медленно до устойчивой сине-сиреневой окраски.
12. ПОСЛЕДОВАТЕЛЬНОЕ ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ КАЛЬЦИЯ И МАГНИЯ
Раздельное определение массовой доли кальция и магния в одной пробе основано на том, что в сильно щелочной среде (рН ≈ 12 ÷ 13), создаваемой введением едкого натра, магний осаждается в виде гидроксида, а кальций остается в растворе и оттитровывается трилоном Б. После определения кальция введением серной или соляной кислоты понижают значение рН раствора до 8 - 9. При этом магний опять переходит в раствор. Затем добавляют аммиачный буферный раствор и определяют массовую долю магния титрованием трилоном Б. Когда общая массовая доля кальция и магния невелика, определять отдельно массовую долю кальция и магния нецелесообразно, так как это не имеет существенного значения, а результаты определения могут иметь значительную погрешность.
Чтобы устранить влияние меди, цинка, железа, марганца, никеля, в пробу исходного раствора накипи вводят диэтилдитиокарбамат натрия, как рекомендовано в п. 11. Присутствие алюминия не мешает определению массовой доли кальция. Влияние алюминия на определение массовой доли магния аналогично влиянию его при определении общей жесткости и также устраняется введением триэтаноламина (см. п. 11).
Реактивы:
Трилон Б, 0,05-молярный или 0,005-молярный раствор.
Едкий натр, 2-нормальный раствор.
Соляная кислота, 2-нормальный раствор.
Серная кислота, 2-нормальный раствор.
Соляная кислота, раствор 1:1.
Аммиачный буферный раствор (см. п. 11).
Индикатор кислотный хром темно-синий (см. п. 11).
Диэтилдитиокарбамат натрия кристаллический.
Триэтаноламин, 30 %-ный раствор.
Выполнение определения:
отбирают в коническую колбу 5 - 10 см3 исходного раствора отложений (а если требуется, то больший объем) и обрабатывают пробу диэтилдитиокарбаматом натрия, как указано в п. 11.
Проверяют рН полученного раствора (по индикаторной бумаге), доводят его до значения 8 - 9, добавляя, если требуется, едкий натр, затем прибавляют 5 см3 2-нормального раствора едкого натра, пробу перемешивают и выжидают около 5 мин для полного осаждения гидроксида магния. Вводят 5 - 7 капель индикатора хром темно-синего, который во избежание его адсорбции гидроксидом магния необходимо добавлять непосредственно перед титрованием. Титруют пробу раствором трилона Б до изменения окраски жидкости из розово-красной в фиолетово-голубую. Титрование заканчивают при отчетливом изменении цвета раствора, не обращая внимания на возможное последующее появление розового оттенка раствора вследствие медленного взаимодействия индикатора с гидроокисью магния. Расход трилона Б соответствует массовой доле кальция.
Для определения массовой доли магния к тому же анализируемому раствору добавляют 2-нормальную соляную кислоту, чтобы нейтрализовать введенные в пробу 5 см3 2-нормаяьного раствора едкого натра и понизить значение рН до 8 - 9, и выжидают 5 мин для растворения гидрата окиси магния. Если присутствует магний, раствор опять приобретает красную окраску. Затем вводят 5 см3 аммиачного раствора и титруют трилоном Б той же концентрации, что и для определения кальция, до изменения окраски анализируемого раствора в сине-сиреневую. При наличии фосфатов титрование нужно производить медленно.
Если переход окраски нечеткий и остается розовый оттенок или предыдущими анализами установлено, что в анализируемом растворе содержится алюминий в количестве, которое мешает определению магния, то перед добавлением аммиачного буферного раствора прибавляют 20 см3 раствора триэтаноламина, затем добавляют аммиачный буферный раствор и титруют трилоном Б.
Массовую долю кальция и магния в пересчете на их оксиды в процентах вычисляют по формулам
где CaO - массовая доля кальция в отложении, %;
MgO - массовая доля магния, %;
a1 - расход раствора три лона Б на титрование кальция, см3;
a2 - расход раствора трилона Б на титрование магния, см3;
М - молярность раствора трилона Б;
К - поправочный коэффициент к данной молярности;
56,08 - молекулярная масса;
40,32 - молекулярная масса MgO.
Определение массовой доли кальция и магния из исходного раствора накипи без отделения других компонентов, входящих в состав исходного раствора, можно выполнить с помощью фотометрического титрования на автоматическом титраторе Т-107 или Т-108.
13. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ФОСФАТОВ
Содержание фосфатов в исходном растворе отложения может быть определено объемным или колориметрическим методом [1 - 3]. Колориметрический метод определения фосфатов менее трудоемкий и выполняется быстрее. Поэтому в лабораториях, где имеются фотоколориметры, предпочтительно определять фосфаты колориметрическим методом.
Определение массовой доли фосфатов колориметрическим методом
Колориметрический метод определения, массовой доли фосфатов основан на образовании комплексной фосфорномолибденовой кислоты, имеющей желтую окраску, которая в присутствии восстановителей превращается в соединение синего цвета. В нижеприведенном методе восстановление производится смесью метола с метабисульфитом калия. Как для получения фосфорномолибденового комплекса, так и для его восстановления имеет существенное значение соблюдение требуемого рН. Получение фосфорномолибдата и его восстановление следует проводить в растворе серной соляной) кислоты с кислотностью до 0,6 - нормальной.
Учитывая, что исходный раствор отложений часто содержит относительно большое количество железа и меди, объем пробы исходного раствора, отбираемого для колориметрического определения фосфатов по приведенному ниже методу, ограничен 20 см3.
Реактивы:
Лимонная кислота, 1-водная, квалификации «х.ч.», 10 %-ный раствор. Раствор пригоден до тех пор, пока в нем не появится осадок.
Восстанавливающий раствор, приготавливаемый из расчета: 20 г метола марки А и 120 г метабисульфита калия растворяют в 300 - 350 см3 дистиллированной воды при температуре примерно 40 °С. Если полученный раствор мутный, его фильтруют в мерную колбу вместимостью 1 дм3, доливают дистиллированной водой до метки и перемешивают. Срок годности раствора не более двух недель.
Раствор молибдата аммония: к 500 см3 дистиллированной воды добавляют 50 см3 концентрированной серной кислоты, после охлаждения прибавляют 50 г измельченного молибдата аммония и, когда он полностью растворится, доводят объем до 1 дм3 дистиллированной водой. Срок годности раствора не более 1 мес.
Стандартный раствор фосфата калия, содержащий 10 мг/дм3 РO3-4. Сначала готовят запасной раствор, содержащий 100 мг/дм3 РO3-4. Для этого в мерной литровой колбе растворяют в дистиллированной воде 0,143 г однозамещенного фосфата калия, предварительно выдержанного в эксикаторе над серной кислотой в течение 1 сут, доливают в колбу дистиллированной воды до метки и тщательно перемешивают.
Стандартный раствор готовят разведением запасного раствора в 10 раз. Стандартный раствор пригоден только в день приготовления.
Построение калибровочного графика:
в ряд мерных колб вместимостью 50 см3 вводят различные количества стандартного раствора фосфата калия, содержащего 10 мг/дм3 РO3-4 (например 1; 2; 3; 5; 10; 15 и 20 см3, что соответствует 0,01; 0,02; 0,03; 0,05; 0,10; 0,15 и 0,20 мг РO3-4.
Объем раствора в каждой колбе доводят до 40 см3 дистиллированной водой. В одну колбу вводят только 40 см3 дистиллированной воды (нулевая проба). Растворы нагревают на водяной бане до 40 - 60 °С, после чего в каждую колбу вводят 0,5 см3 раствора лимонной кислоты, 2 см3 восстанавливающего раствора и перемешивают, затем добавляют 2 см3 раствора молибдата аммония и опять перемешивают. Спустя 10 мин добавляют в колбы дистиллированной воды до метки, растворы перемешивают и затем колориметрируют. В качестве раствора для сравнения используют дистиллированную воду со всеми реактивами. Колориметрирование проводят с красными светофильтрами при длине волны 813 нм или возможно более близкой к ней.
По результатам колориметрирования строят график, откладывая по оси абсцисс содержание РO3-4 в пробе (мг), а по оси ординат соответствующие показания оптической плотности.
Выполнение определения:
в мерную колбу вместимостью 50 см3 отбирают в зависимости от предполагаемого содержания фосфатов 0,5 - 20 см3 исходного раствора отложения, добавляют дистиллированной воды до 40 см3 нагревают на водяной бане до 40 - 60 °С. Затем вводят 0,5 см3 раствора лимонной кислоты, 2 см3 восстанавливающего раствора, содержимое колбы перемешивают, добавляют 2 см3 раствора молибдата аммония и вновь перемешивают.
Спустя 10 мин доливает в колбу до метки дистиллированной воды, а затем колориметрируют.
В качестве раствора сравнения используют дистиллированную воду со всеми реактивами.
Для колориметрирования применяют те же кюветы и светофильтр, что и при построении калибровочного графика, по которому определяют содержание фосфатов в пробе.
Массовую долю фосфатов в пересчете на P2O5 вычисляют по формуле, %
где a - массовая доля РO3-4 в пробе, мг;
0,747 - коэффициент пересчета РO3-4 в P2O5.
Определение массовой доли фосфатов объемным методом
Сущность метода состоит в том, что фосфаты осаждают молибденовокислым аммонием из исходного раствора отложения, подкисленного азотной кислотой, в присутствии азотнокислого аммония, который прибавляют для ускорения осаждения фосфатов и уменьшения растворимости осадка (действие общего иона). Осадок фосфорномолибдата отфильтровывают, отмывают и растворяют в титрованном растворе едкого натра, добавляемого в избытке.
При этом происходит реакция, которая может быть выражена уравнением
(NH4)3H4[P(Mo2O7)6] + 23NaOH = 11Na2MoO4 + (NH4)2MoO4 + NaNH4HPO4 + 13H2O
Избыток едкого натра оттитровывают кислотой. Массовую долю фосфатов вычисляют по расходу щелочи на их растворение.
Реактивы:
Молибденовокислый аммоний, 3 %-ный раствор.
Азотнокислый аммоний, 34 %-ный раствор.
Азотная кислота, 25 %-ный раствор.
Азотнокислый калий, 25 %-ный раствор (для промывания употребляют 1 %-ный раствор, который готовят разбавлением 25 %-ного раствора).
Едкий натр, 0,1-нормальный раствор.
Раствор соляной или серной кислоты, 0,1-нормальной концентрации.
Метилоранж, 0,1 %-ный раствор.
Фенолфталеин, 1 %-ный спиртовый раствор.
Выполнение определения:
отбирают в химический стакан 25 или 50 см3 исходного раствора отложения, приливают 20 см3 раствора азотнокислого аммония и 10 см3 азотной кислоты, нагревают до кипения и вливают (по стеклянной палочке) в кипящую жидкость 20 см3 нагретого до 50 °С раствора молибденовокислого аммония. Сразу же прекращают нагревание жидкости, перемешивают ее вращением стакана (пользоваться стеклянной палочкой для перемешивания не рекомендуется) и оставляют на 2 - 3 ч. Выпавший в осадок фосфоромолибдат должен быть чисто-желтого цвета; «белесый» оттенок свидетельствует о неправильном осаждении и образовании молибденовой кислоты. В этом случае необходимо повторить осаждение, взяв новую порцию исходного раствора или растворив осадок постепенным добавлением 25 %-ного раствора аммиака, затем нагреть жидкость до кипения и добавить в нее малыми порциями 15 см3 25 %-ного раствора азотной кислоты. После 2 - 3-часовой выдержки осадок отфильтровывают через два фильтра «синяя лента», обмывают стенки стакана и промывают осадок 1 %-ным раствором нитрата калия до нейтральной реакции промывных вод по метилоранжу.
Для проверки полноты отмывания осадка и фильтра от кислоты собирают в стаканчик или коническую колбу 10 см3 фильтрата, вводят 1 - 2 капли метилоранжа и одну каплю 0,1-нормального раствора щелочи. Если жидкость окрасится в желтый цвет, то промывание считают законченным.
Отмытый осадок вместе с фильтром помещают в стакан, в котором проводилось осаждение, приливают 25 - 30 см3 горячей дистиллированной воды и столько же 0,1-нормального раствора едкого натра и взбалтывают до полного растворения желтого осадка (при необходимости добавляют щелочь). После этого вводят 2 - 3 капли фенолфталеина и оттитровывают избыток щелочи раствором кислоты, титруя окрашенную в красный цвет жидкость до исчезновения окраски. Содержание фосфатов в пересчете на P2O5 вычисляют по формуле, %
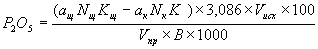 (
(где ащ, ак - объемы растворов щелочи и кислоты, израсходованных при выполнении анализа, см3;
Nщ, Nk - нормальность растворов щелочи и кислоты;
Kщ, Kk - поправочные коэффициенты растворов щелочи и кислоты к данной нормальности;
3,086 - эквивалентная масса P2O5 в реакции со щелочью.
14. ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ СУЛЬФАТОВ
Массовую долю сульфатов определяют массовым или объемным методом. Сущность нижеприведенного объемного метода заключается в осаждении сульфата бария из исходного раствора, отделении и промывке осадка, растворении осадка в аммиачной среде титрованным раствором трилона Б, избыток которого оттитровывают раствором соли магния. По количеству трилона Б, израсходованного на растворение сернокислого бария, определяют содержание сульфатов в анализируемой пробе [14].
Реактивы:
Трилон Б, 0,05-молярный раствор.
Барий хлористый, 0,05-молярный (0,1-нормальный) раствор. Для приготовления его растворяют 12,2 г BaCl2 ´ 2H2O в 1 дм3 дистиллированной воды.
Магний хлористый, 0,05-можярный (0,1-нормальный) раствор. Приготавливают из фиксанола или 10,17 г MgCl2 ´ 6H2O растворяют в дистиллированной воде, доводят объем до 1 дм и устанавливают титр по титрованному раствору трилона.
Аммиачный буферный раствор 2 %-ный по NH4Cl и по NH3 (см. п. 11).
Аммиак, 9-нормальный раствор. 67,5 см3 концентрированного (25 %-ного) аммиака разбавляют дистиллированной водой до 100 см3. Раствор индикатора кислотного хром темно-синего (см. п. 11).
Выполнение определения:
исходя из предполагаемого содержания сульфатов, в коническую колбу вместимостью 500 см3 отбирают такой объем исходного раствора отложений, чтобы в нем содержалось не более 20 мг SO2-4.
Для выбора объема пробы к 5 - 10 см3 исходного раствора прибавляют 3 - 5 капель раствора хлористого бария и нагревам до кипения. При отсутствии или появлении следов белой мути отбирают 50 - 100 см3 исходного раствора, а при значительном помутнении и выпадении осадка отбирают меньший объем пробы.
К отобранной пробе исходного раствора добавляют дистиллированной воды до 100 см3 и подкисляют соляной кислотой до рН ≈ 1, прибавляют 10 - 15 см3 раствора хлористого бария, кипятят 10 мин и оставляют на водяной бане или в теплом месте на 1 - 2 ч. Затем пробу фильтруют через бензольный фильтр «синяя лента», предварительно запаренный кипящей дистиллированной водой, промывают колбу с осадком несколько раз горячей дистиллированной водой, не удаляя приставшего к стенкам осадка, сливая промывные воды на фильтр с осадком. Потом фильтр с осадком промывают еще 3 - 4 раза горячей водой, проверяют на полноту промывания разбавленной серной кислотой и переносят в ту же колбу, в которой производилось осаждение, прибавляют 10 - 15 см3 9-нормального раствора аммиака, разворачивают фильтр стеклянной палочкой на дне колбы, прибавляют 10 - 15 см3 0,05-молярного раствора трилона Б и доводят объем до 100 см3, а при повышенном содержании сульфатов до 200 см3. При недостаточном разбавлении раствора могут получиться заниженные результаты анализа из-за неполного растворения сульфата бария, даже при значительном избытке трилона Б и поддержании требуемой щелочной среды рН ³ 10. Содержимое колбы кипятят 10 - 15 мин до полного растворения осадка, периодически проверяя (по универсальной индикаторной бумаге) рН раствора, которое должно быть не ниже 10, так как сульфат бария растворяется трилоном Б только в щелочной среде. При уменьшении рН из-за улетучивания аммиака добавляют еще 5 см3 раствора аммиака.
После растворения осадка раствор охлаждают, вводят соответственно 5 - 10 см3 аммиачного буферного раствора, 5 - 10 капель индикатора кислотного хром темно-синего и титруют избыток трилона Б раствором соли магния до изменения окраски из голубой в розовую.
Массовую долю сульфатов в пересчете на SO3 в процентах вычисляют по формуле
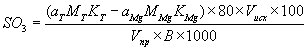 (
(где аT аMg - объемы растворов трилона Б и соли магния, израсходованных при выполнении анализа, см3;
MT, MMg - молярность растворов трилона и соли магния;
KT, KMg - поправочные коэффициенты растворов трилона и соли магния к данной молярности;
80 - молекулярная масса SO3 в реакции с трилоном Б.
Список использованной литературы
1. Инструкция по анализу воды, пара и отложений в теплосиловом хозяйстве. - М.: Энергия, 1967.
2. Методика контроля состояния оборудования; определение количества и состава отложений. - М.: СПО ОРГРЭС, 1976.
3. Методика анализа накипей и отложений. - М.-Л.: Энергия, 1966.
4. Информационное письмо № 6-82. Ускоренный анализ водорастворимой части отложений. - М.: СПО Союзтехэнерго, 1982.
5. ГОСТ 10538.1-72. Метод определения содержания двуокиси кремния в золе.
6. Воскресенский П.И. Техника лабораторных работ. М.: Химия, 1973.
7. Пятницкий И.В., Симоненко В.И., Ковальчук Л.И. Ускоренные методы фотометрического определения железа в медных сплавах. Заводская лаборатория, 1982, № 3.
8. Методика химического контроля при очистках теплообменников из латуни и медноникелевых сплавов смесью низкомолекулярных кислот и смесью их с соляной кислотой. М.: СПО Союзтехэнерго, 1980.
9. Информационное письмо № 10-80. Ускоренное определение содержания меди, цинка и марганца в отложениях, образующихся на внутренних поверхностях нагрева теплосилового оборудования. М.: СПО Союзтехэнерго, 1980.
10. Файнберг С.С., Филлипова А.А. Анализ руд цветных металлов. М.: Металлургиздат, 1963.
11. Лурье Ю.Ю. Аналитическая химия промышленных сточных вод. М.: Химия, 1984.
СОДЕРЖАНИЕ