 | Информационная система | |
|
УТВЕРЖДАЮ Член коллегии Минхиммаша А.М. Васильев «8» сентября 1980 г. |
РУКОВОДЯЩИЕ ТЕХНИЧЕСКИЕ МАТЕРИАЛЫ
|
УСКОРЕННЫЕ И МАРКИРОВОЧНЫЕ МЕТОДЫ |
РД РТМ
20-362-80 - Взамен РТМ
2631-70 - |
|
Письмом Министерства химического и нефтяного машиностроения от 08.09 1980 г № 11-10-4/1601 |
|
от 08.09 1980 г № 11-10-4/1601 срок введения установлен
с 01.10.1980 г.
Настоящие руководящие технические материалы распространяются на химические и физические метода исследования химсостава основных и сварочных материалов, применяемых в химическом и нефтяном машиностроении (кроме защитных газов).
Устанавливают типовые методы исследования материалов, имеющих различную основу, метода подсчета результатов и технику безопасности.
Рекомендуются к применению в ЦЗЛ, как сборник методических инструкций по проведению химического и спектрального анализа чугунов, сталей и др. материалов.
РУКОВОДЯЩИЙ ТЕХНИЧЕСКИЙ МАТЕРИАЛ
УСКОРЕННЫЕ И МАРКИРОВОЧНЫЕ МЕТОДЫ
ХИМИЧЕСКОГО
И СПЕКТРАЛЬНОГО АНАЛИЗА ОСНОВНЫХ И СВАРОЧНЫХ
МАТЕРИАЛОВ В ХИМНЕФТЕАППАРАТОСТРОЕНИИ
МЕТОДЫ ХИМИЧЕСКОГО АНАЛИЗА ЧУГУНОВ И СТАЛЕЙ
РД РТМ 26-362 80
Настоящий руководящий технический материал распространяется на методы исследования химсостава материалов, применяемых при изготовлении нефтезаводской и химической аппаратуры: чугунов, низколегированных и высоколегированных сталей, сварочных проволок, наплавленного металла, а так же металла сварных швов на основные маркировочные и легирующие элементы.
Методы анализа элементов, указанные в приложении, являются ускоренными по сравнению с методами рекомендуемыми ГОСТами, перечисленными на стр. 156.
Допускаемые расхождения результатов анализа не превышают величин установленных указанными ГОСТами.
1. ОБЩИЕ ТРЕБОВАНИЯ К МЕТОДАМ АНАЛИЗА
1.1. Проба стали для химического и спектрального анализов должна быть подготовлена по ГОСТ 7565-73.
Проба чугуна для химического анализа должна быть подготовлена по ГОСТ 805-69 и по ГОСТ 4832-72 и иметь однородную и мелкозернистую структуру.
Проба наплавленного электродом металла для химического анализа должна быть подготовлена по ГОСТ 9466-75 и ГОСТ 7122-75.
Пробы для анализа других материалов готовят в соответствии с требованиями ГОСТов на поставку.
1.2. Все применяемые реактивы должны иметь степень чистоты «химически чистый» (х.ч.) или «особой чистоты» (ос.ч.). Применение реактивов степени, чистоты «чистый для анализа» (ч.д.а.) допускается только в том случае, если промышленность не выпускает реактивов указанной выше степени чистоты.
1.3. Для приготовления растворов и при проведении анализов применяют дистиллированную воду по ГОСТ 6709-72.
1.4. В выражении «разбавленная 1:1, 1:2» и т.д. первые цифры означают объемные части разбавляемого реактива (например, концентрированной кислоты), вторые - объемные части используемого для разбавления растворителя (вода).
1.5. Концентрация растворов выражается в процентах; число процентов указывает массовую долю растворенного вещества в граммах, в 100 г раствора.
1.6. Анализируемую пробу взвешивают на аналитических весах с погрешностью не более 0,0002 г.
1.7. Перед анализом необходимо проверять градуировку применяемой мерной посуды (пипеток, бюреток, мерных колб и пр.), а также термометров.
1.8. При каждой серии определений необходимо проводить не менее двух контрольных опытов в условиях анализа для внесения в результат определения поправки на загрязнение реактивов.
1.9. При титриметрическом анализе титр раствора устанавливают не менее, чем по трем навескам исходного вещества или стандартного образца.
1.10. Проведение анализа фотоколориметрическим методом.
1.10.1. Аликвотная часть раствора и размер кюветы должны обеспечить оптимальные условия определения заданного интервала концентраций элемента.
1.10.2. Массовую долю определяемого элемента в анализируемой пробе находят по градуировочному графику, на оси абсцисс которого откладывают массовую долю определяемого элемента в процентах, а на оси ординат - оптическую плотность раствора окрашенного комплексного соединения. Можно также использовать метод сравнения оптической плотности пробы с оптической плотностью стандартного раствора определяемого элемента или раствора стандартного образца близкого по составу к анализируемой пробе.
1.10.3. Градуировочные графики, построенные по стандартным растворам, обязательно проверяют по 1 - 2 стандартным образцам.
1.10.4. Водные растворы реактивов предварительно фильтруют.
1.10.5. Массовую долю элементов при химическом анализе определяют по трем параллельным навескам, при спектральном анализе по трем параллельным определениям (по три измерения в каждом) или двум параллельным определениям (по три измерения в каждом), если расхождение при анализе не превышает допускаемых, приведенных в соответствующем стандарте. Одновременно в тех же условиях анализируют стандартный образец, химический состав которого соответствует анализируемой пробе, при этом массовая доля контролируемого элемента в стандартном образце и в анализируемой пробе не должны различаться более чем в два раза.
Средний результат анализа стандартного образца не должен отличаться от результата, указанного в свидетельстве, более чем на половину максимальной величины абсолютных допускаемых расхождений для трех параллельных определений.
2. КУЛОНОМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ ОБЩЕГО УГЛЕРОДА
2.1. Сущность метода
Навеску чугуна или стали сжигают в печи в токе кислорода при температуре от 1200 до 1250 °C. Образующийся углекислый газ поглощается раствором и вызывает повышение кислотности, что в свою очередь приводит к изменению э.д.с. индикаторной системы pH-метра.
Количество электричества, необходимое для достижения первоначального значения pH поглотительного раствора и пропорциональное концентрации углерода в пробе, фиксируется специальным кулонометром - интегратором тока, показывающим непосредственное количество углерода в пробе (в процентах).
2.2. Аппаратура, реактивы и растворы. Экспресс-анализатор углерода типа АН-29, АН-160.
Схема экспресс-анализатора углерода типа AH-160 приведена черт. 1.
Экспресс-анализатор AH-160 состоит из трубчатой электропечи 1, электролитической ячейки 2, поглотительного раствора 3, стеклянного электрода 4, вспомогательного электрода 5, pH-метра 6, регулятора 7, источника генераторного тока 8, катода 9, анода 10, проницаемой для тока перегородки, 11, вспомогательного раствора 12, интегратора тока 13.
Баллон с кислородом по ГОСТ 5583-78, снабженный редукционным вентилем.
Крючок из низкоуглеродистой жаропрочной проволоки диаметром мм 18 - 20, длиной мм 750 - 900, с помощью которого фарфоровые лодочки с пробой помещают в трубку для сжигания и извлекают из нее.
Фарфоровые лодочки, прокаленные в токе кислорода при температуре от 1200 до 1250 °C, хранят в эксикаторе. Шлиф крышки эксикатора не следует покрывать смазывающим веществом.
СХЕМА ЭКСПРЕСС-АНАЛИЗАТОРА СТАЛИ НА УГЛЕРОД
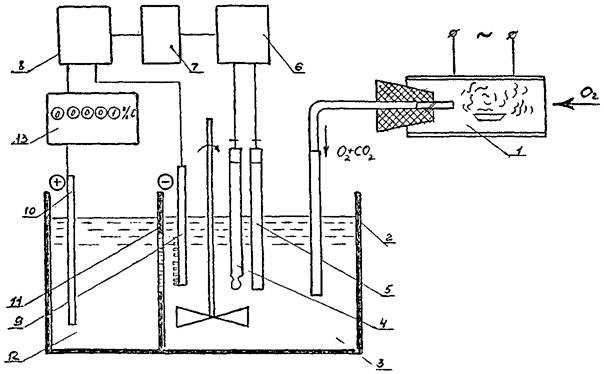
Черт. 1
Аскарит.
Поглотительный раствор; готовят следующим образом: 50 г хлористого калия по ГОСТ 4234-77, 50 г хлористого стронция по ГОСТ 4140-74 помещают в мерную колбу вместимостью один литр, прибавляют около 500 см3 воды и после растворения разбавляют водой до метки.
Вспомогательный раствор; готовят следующим образом: 50 г хлористого калия по ГОСТ 4234-77, 50 г ферроцианида калия по ГОСТ 4207-75 и 1,0 г буры помещают в мерную колбу вместимостью один литр, прибавляют около 500 см3 воды и после растворения разбавляют водой до метки.
Свинец гранулированный; плавень готовят следующим образом: гранулы свинца промывают в ацетоне и высушивают, разбивают или раскатывают в тонкие пластинки, все поверхности, с которыми соприкасается свинец, обезжиривают, нарезают мелкие отрезки и еще раз промывают в ацетоне, сушат и хранят в банке с притертой пробкой или боксе.
2.3. Проведение анализа
Прибор подготавливают к работе в соответствии с инструкцией.
Навеску стали массой 0,5 г переносят в фарфоровую лодочку, покрывают равномерным слоем плавня свинца в количестве 1,0 г, лодочку с помощью крючка вводят по направлению движения кислорода в предварительно разогретую до 1250 °C в печи фарфоровую трубку и устанавливают в центральной, наиболее накаленной части трубки для сжигания, которую сразу же закрывают затвором.
Нажатием на кнопку «сброс» устанавливают показание индикаторного цифрового табло на «нуль». После полного сгорания стружки с плавнем (2 мин), о чем судят по завершению процесса титрования, записывают результат анализа по показанию табло, открывают затвор и извлекают лодочку.
В условиях анализа определяют массовую долю углерода в свинце гранулированном (плавне). Для этого в лодочку помещают навеску свинца массой 1,0 г, сжигают в печи и полученный результат вычитают из показания каждой пробы.
2.4. Обработка результатов
2.4.1. Массовая доля углерода в процентах при навеске массой 0,5 г соответствует показанию цифрового табло прибора с учетом контрольной пробы.
2.4.2. Абсолютные допускаемые расхождения между результатами двух параллельных определений не должны превышать значений указанных (в табл. 1).
Таблица 1
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,01 до 0,02 |
0,005 |
|
Св. 0,02 " 0,08 |
0,010 |
|
" 0,08 " 0,20 |
0,020 |
|
" 0,20 " 0,50 |
0,030 |
|
" 0,50 " 1,0 |
0,040 |
|
" 1,0 " 2,5 |
0,05 |
|
" 2,5 " 5,0 |
0,10 |
3. ГАЗООБМЕННЫЙ МЕТОД ОПРЕДЕЛЕНИЯ УГЛЕРОДА
3.1. Сущность метода
Метод основан на сжигании навески стали или чугуна в токе кислорода при температуре от 1200 до 1250 °C с последующим поглощением образовавшегося углекислого газа раствором едкого калия.
Массовую долю углерода определяют по разности между первоначальным объемом газов и объемом, полученным после поглощения углекислого газа раствором едкого кали.
3.2. Аппаратура, реактивы и растворы
Схема установки для определения углерода приведена на черт. 2. Установка состоит из следующих элементов: кислородного баллона, снабженного редукционным вентилем для регулирования скорости поступления кислорода в печь 1; промывной склянки Тищенко с 20 %-ным раствором едкого кали 2; промывной склянки Тищенко с серной кислотой 3; горизонтальной трубчатой печи 4 с силитовыми нагревателями, обеспечивающей нагрев до температуры не ниже 1250 °C; шарика-фильтра, заполненного хлопчато-бумажной ватой 7; термопары с прибором, обеспечивающим поддержание необходимой температуры 8; змеевикового холодильника для охлаждения поступающих из печи двуокиси углерода с кислородом 9; газоизмерительной бюретки-эвдиометра 10, снабженного термометром 11, укрепленным в верхней расширенной части (в верхней части эвдиометра имеется пустотелый поплавок, поднимающийся при заполнении бюретки жидкостью и запирающий верхнее отверстие); подвижной шкалы 12, прикрепленной к узкой части бюретки, при помощи которой измеряют объемы газов (шкала соответствует только той бюретке, к которой она прилагается, перенос ее с одной бюретки на другую не допускается); уравнительной склянки 13 для перекачивания газовой смеси из бюретки в поглотительный сосуд (в уравнительную склянку наливают воду, от 5 до 6 капель серной кислоты и несколько капель раствора метилового красного); поглотительного сосуда, наполненного 40 %-ным раствором едкого калия; фарфоровой неглазурованной трубки 6, газонепроницаемой, длиной мм 750 - 800, с внутренним диаметром мм 18 - 20, концы которой должны выступать из печи не менее чем на 200 мм с каждой стороны; лодочки фарфоровой 5 неглазурованной по ГОСТ 6675-73; крючок из низкоуглеродистой жаропрочной проволоки диаметром мм 3 - 5, длиной мм 500 - 600, с помощью которого лодочку с пробой помещают в трубку для сжигания и извлекают из нее.
СХЕМА УСТАНОВКИ ДЛЯ ОПРЕДЕЛЕНИЯ УГЛЕРОДА
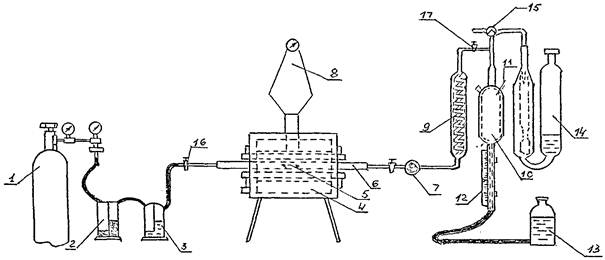
Черт. 2
Плавни:
Медь (П) окись, порошок по ГОСТ 4469-48.
Ванадий (У) окись, по ТУ 6-09-4093-78.
Свинец гранулированный, по МРТУ 6-09-4030-67.
Железо, порошок, низкоуглеродистое.
Во всех веществах, применяемых в качестве плавней, проверяют массовую долю в них углерода, которая не должна превышать 0,002 %.
3.3. Проведение анализа
Перед началом работы проверяют герметичность аппарата. Аппарат герметичен, если уровни растворов в поглотителе и эвдиометре не изменяются при закрытых кранах 16 и 15, а кран 17 находится в положении соединения трубки для сжигания с мерной бюреткой.
Навеску обезжиренной стружки стали массой от 0,5 до 1,0 г или 0,2 г чугуна помещают в заранее прокаленную фарфоровую лодочку, прибавляют от 0,5 до 1,0 г плавня и с помощью «крючка» вдвигают лодочку в трубку для сжигания, которая помещена в предварительно нагретую до температуры не ниже 1250 °C трубчатую печь. Лодочку устанавливают в наиболее накаленной части трубки и тот час же закрывают отверстие трубки пробкой с резиновым шлангом.
По истечении одной минуты (время необходимое для равномерного прогрева стружки, плавня и лодочки до температуры печи), в трубку для сжигания начинают подавать кислород со скоростью 4 - 5 пузырьков в секунду.
Одновременно с подачей кислорода трехходовой кран 17 ставят в положение, при котором газовая смесь кислорода и углекислого газа поступает из печи через змеевиковый холодильник в эвдиометр.
Двухходовой кран 15 полностью перекрыт. Как только эвдиометр полностью заполняется газом и уровень жидкости достигнет нулевого деления шкалы, трехходовой кран 17 ставят в положение, при котором газ из печи выходит в атмосферу, а с помощью крана 15 эвдиометр соединяют с атмосферой, уравнительной склянкой выравнивают положение уровней жидкости в склянке и в бюретке. Выждав от 15 до 20 секунд для полного отекания жидкости со стенок эвдиометра, на том же уровне устанавливают нуль подвижной шкалы. Эвдиометр с помощью крана 15 соединяют с поглотительным сосудом для поглощения углекислого газа из газовой смеси. Для этого газовую смесь (кислород с углекислым газом) с помощью уравнительной склянки переводят в поглотитель, затем обратно в эвдиометр, повторяя эту операцию не менее двух раз. Оставшийся газ (кислород) переводят в газоизмерительную бюретку, разобщают ее с поглотителем краном - 15, который ставят в положение соединения с атмосферой и производят замер уровня жидкости, записывают соответствующее деление шкалы. Одновременно производят замер температуры и давления атмосферы. Наполняют эвдиометр жидкостью из уравнительной склянки, закрывают кран - 15 и готовят аппарат для следующего сжигания.
3.4. Обработка результатов
3.4.1. Массовую долю углерода (X) в процентах вычисляют по формуле
![]()
где A и A1 - показания шкалы эвдиометра после поглощения углекислого газа при сжигании навесок анализируемого образца и контрольной пробы соответственно;
K - поправочный коэффициент на температуру и давление по таблицам, прилагаемым к прибору;
m - масса навески образца, г.
3.4.2. Абсолютные допускаемые расхождения между результатами трех параллельных определений не должны превышать значений указанных (в табл. 2).
Таблица 2
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,01 до 0,02 |
0,005 |
|
Св. 0,02 " 0,08 |
0,010 |
|
" 0,08 " 0,20 |
0,020 |
|
" 0,20 " 0,50 |
0,030 |
|
" 0,50 " 1,00 |
0,040 |
|
" 1,00 " 2,5 |
0,05 |
|
" 2,5 " 5,0 |
0,10 |
4. ОБЪЕМНЫЙ МЕТОД ОПРЕДЕЛЕНИЯ СЕРЫ
4.1. Сущность метода
Метод основан на сжигании навески стали или чугуна в токе кислорода при температуре от 1200 до 1250 °C с поглощением образующейся двуокиси серы водой и титрования сернистой кислоты раствором йода или смесью йодноватокислого и йодистого калия в присутствии индикатора крахмала.
4.2. Аппаратура, реактивы и растворы
Схема установки для определения серы приведена на черт. 3.
Установка для газообъемного определения содержания серы состоит из следующих элементов: баллона с кислородом 1; редукционного вентиля для регулирования скорости поступления кислорода 2; трубчатой горизонтальной электропечи 7, обеспечивающей температуру нагрева не ниже 1250 °C; газонепроницаемой трубки 9 из неглазурованного фарфора, внутренним диаметром мм 18 - 20 и длиной мм 750 - 900. Концы трубок должны выступать наружу из печи не менее чем на 200 - 250 мм. Трубку закрывают с двух концов плотно пригнанными резиновыми пробками. В отверстия пробок вставляют стеклянные трубки диаметром 5 мм, лодочек фарфоровых 6 неглазурованных № 1 и № 2 по ГОСТ 6675-73, предварительно прокаленных в токе кислорода при рабочей температуре; термопары с терморегулятором или гольванометром 8; шарика, заполненного стеклянной или хлопчатобумажной ватой 10, для улавливания окислов железа и др., образующихся при сжигании навески; промывной склянки с серной кислотой 4, промывной склянки с 40 %-ным раствором едкого калия 3; поглотительного сосуда 11, внутренним диаметром от 35 до 40 мм и высотой 150 мм, наполненного до половины дистиллированной водой с крахмалом и несколькими каплями йода, чтобы раствор окрасился в бледно-голубой цвет. Внизу поглотительный сосуд оканчивается стеклянным краном 13, необходимым для слива оттитрованного раствора; бюретки 14, расположенной над поглотительным сосудом; сосуда с раствором для сравнения окраски. Плавни - олово по ГОСТ 546-67, медь металлическая, предварительно проверенные в условиях анализа на содержание серы.
СХЕМА УСТАНОВКИ ДЛЯ ОПРЕДЕЛЕНИЯ СЕРЫ
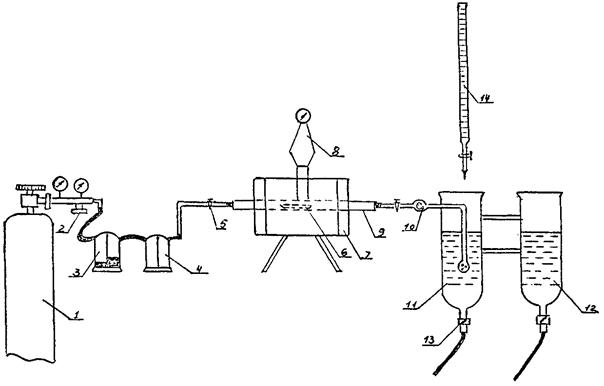
Черт. 3
Крючок, с помощью которого лодочки с пробой помещают в фарфоровую трубку и извлекают из нее, изготовленный из жаропрочной низкоуглеродистой проволоки диаметром мм 3 - 5 и длиной мм 500 - 600.
Калия гидрат окиси (едкое кали), 40 %-ный раствор.
Кислота серная по ГОСТ 4204-77.
Крахмал растворимый по ГОСТ 10163-76; готовят следующим образом: 5 г растворимого крахмала растирают в фарфоровой ступке с 50 см3 воды и тонкой струйкой вливают в мерную колбу вместимостью 2 литра, с водой нагретой до кипения, доводят водой до метки, перемешивают.
Йод кристаллический по ГОСТ 4159-79, титрованный раствор; готовят следующим образом: 1 г йода взвешивают в стаканчике с притертой пробкой, пересыпают в колбу с 5 г йодистого калия по ГОСТ 4232-74 и 50 см3 воды, после полного растворения йода раствор переводят в мерную колбу вместимостью 3 литра, доводят до метки водой и перемешивают. Раствор хранят в склянке из темного стекла.
Калий иодистый по ГОСТ 4232-74.
Титр раствора йода устанавливают по стандартным образцам с известной массовой долей серы и химическим составом, близким по химсоставу к анализируемой пробе. Сжигание навесок стандартных образцов проводят в тех же условиях, что и анализ.
Титр раствора (Т), выраженный в г/см3, вычисляют по формуле
![]()
где C - массовая доля серы в стандартном образце, %;
m - масса навески стандартного образца, г;
V - объем раствора йода, израсходованный на титрование стандартного образца, см3;
V1 - объем раствора йода, израсходованный на титрование контрольной пробы, см3.
4.3. Подготовка аппаратуры
Перед началом работы установку проверяют на герметичность.
В трубке и лодочке для сжигания пробы определяют наличие летучих (восстановительных веществ). Для этого, по достижении в печи температуры от 1200 до 1250 °C, трубку закрывают с обеих сторон пробками, наливают в оба сосуда от 110 до 120 см3 крахмального раствора, приливают из бюретки несколько капель титрованного раствора йода до появления голубой окраски и, открыв кран, пропускают ток кислорода с такой скоростью, чтобы уровень жидкости в поглотительном сосуде поднялся на мм 30, 40.
Обесцвечивание раствора в поглотительном сосуде при пропускании кислорода в течение нескольких минут свидетельствует о выделении из трубки восстановительных газообразцах веществ, реагирующих с йодом. В любом случае, не прекращая подачи кислорода, приливают к поглотительному раствору титрованный раствор йода до тех пор, пока интенсивность окраски растворов в обоих сосудах не станет одинаковой. Для проверки правильности работы установки сжигают от 2 до 3 навесок стандартного образца стали или чугуна в присутствии плавня, затем сжигают навеску плавня для внесения поправки в контрольный опыт.
4.4. Проведение анализа
Навеску стали (чугуна) массой от 0,5 до 1,0 г помещают в лодочку, равномерно распределяют по дну и покрывают равномерным слоем плавня в количестве от 0,5 до 1,0 г. Лодочку с пробой и плавнем при помощи крючка помещают в наиболее нагретую часть фарфоровой трубки и немедленно закрывают трубку резиновой пробкой, в которую вставлена стеклянная трубка для отвода газообразных продуктов сжигания в поглотительный сосуд. Скорость пропускания кислорода составляет 2,5 л/мин.
В процессе горения навески наблюдают за изменением окраски жидкости в поглотительном сосуде, где происходит поглощение окислов серы. Во время сжигания окраска растворов в поглотительном сосуде должна быть все время близкой к окраске раствора сравнения. Для этого к раствору в поглотительном сосуде по мере уменьшения интенсивности окраски добавляют раствор иода до получения одинаковой интенсивности окраски в обоих сосудах. В этом случае титрование считают законченным. Для проверки полноты сгорания навески кислород продолжают подавать еще в течение 1 минуты. Если интенсивность окраски раствора не уменьшится, определение считают законченным, если уменьшится - титрование продолжают. После сжигания пробы лодочку вынимают крючком из печи, поглотительный раствор сливают из сосуда и промывают сосуд водой.
4.5. Обработка результатов
4.5.1. Массовую долю серы (X) в процентах вычисляют по формуле
![]()
где V2 - объем раствора йода, израсходованный на титрование анализируемого образца, см3;
V1 - объем раствора йода, израсходованный на титрование контрольной пробы; см3;
T - титр раствора йода, установленный по стандартным образцам и выраженный в г/см3 серы;
m - масса навески анализируемого образца, г.
4.5.2. Абсолютные допускаемые расхождения между результатами трех параллельных определений не должны превышать значений, указанных (в табл. 3).
Таблица 3
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,002 до 0,004 |
0,0015 |
|
Св. 0,004 " 0,010 |
0,0025 |
|
" 0,010 " 0,025 |
0,004 |
|
" 0,025 " 0,050 |
0,006 |
|
" 0,050 " 0,10 |
0,008 |
|
" 0,10 " 0,25 |
0,012 |
5. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ ФОСФОРА В ЧУГУНАХ, УГЛЕРОДИСТЫХ И НИЗКОЛЕГИРОВАННЫХ СТАЛЯХ
Фотоколориметрический метод определения фосфора основан на образовании растворимой комплексной соли фосфорномолибденовой гетерополикислоты, окрашивающей раствор в интенсивный желтый цвет с последующим восстановлением молибдена в комплексе до синего цвета, так называемой молибденовой сини, которая более чувствительна. Интенсивность окрашивания при соблюдении определенных условий пропорциональна массовой доле фосфора. Чувствительность метода составляет 0,00001 г фосфора в 50 см3 раствора. Фотометрирование проводят с красным светофильтром, в основном в солянокислой среде, при оптимальной кислотности не выше 0,62 Н.
В качестве восстановителя применяют хлористое олово, сернокислый гидразин, сернистокислый натрий и др. Введение больших количеств восстановителя приводит к уменьшению оптической плотности раствора. Введение молибдата аммония в количествах больших, чем это указано в методике, также приводит к изменению диапазона кислотности, в котором светопоглощение стабильно.
Влияние небольших количеств кремния и мышьяка устраняют, восстанавливая фосфоромолибдат при другом режиме кислотности, чем кремнемолибдат или мышьякомолибденовый комплекс. Пятивалентный ванадий восстанавливают насыщенным раствором соли Мора.
Хром окисляют до шестивалентного состояния в сернокислой среде и осаждают фосфор с железом.
5.2. Аппаратура, реактивы и растворы
Фотоколориметр ФЭК-56 или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота азотная по ГОСТ 4461-77, разбавленная 1:1.
Калий марганцовокислый по ГОСТ 20490-75, 4 %-ный раствор.
Натрий сернистокислый (сульфит) по ГОСТ 195-77, 20 %-ный раствор, не содержащий фосфорнокислых солей.
Аммоний молибденовокислый по ГОСТ 3765-78, 5 %-ный раствор.
Кислота соляная по ГОСТ 3118-77, 4 н раствор.
Аммиак водный по ГОСТ 3760-79.
5.3. Проведение анализа
Навеску стали массой 0,3 г помещают в коническую колбу вместимостью 250 см3 и растворяют в 15 см3 азотной кислоты, разбавленной 1:1. Раствор кипятят до полного удаления бурых паров. После растворения навески к кипящему раствору приливают 6 см3 раствора марганцовокислого калия и кипятят до выпадения осадка перекиси марганца и полного исчезновения фиолетовой окраски. Осадок растворяют в 3 см3 раствора сернистокислого натрия и кипятят до полного удаления окислов азота.
К охлажденному раствору добавляют 40 см3 воды, приливают аммиак водный до выпадения нерастворяющегося осадка гидроокиси железа, который затем растворяют в 4 н соляной кислоте, прибавляя последнюю осторожно до полного растворения гидроокисей и избыток 6 см3. Приливают 12,5 см3 раствора сернистокислого натрия, кипятят до обесцвечивания раствора и восстановления железа.
После охлаждения раствора приливают 18 см3 4 н соляной кислоты, переносят в мерную колбу вместимостью 100 см3, доводят до метки водой и перемешивают.
Аликвотную часть раствора 50 см3 помещают в сухую коническую колбу, вместимостью 100 см3, медленно, при постоянном помешивании приливают 4,5 см3 раствора молибденовокислого аммония, выдерживают 10 минут и измеряют оптическую плотность раствора на спектрофотометре при длине волны 670 нм или на фотоколориметре, со светофильтром, имеющим область пропускания в интервале от 650 до 700 нм, в кювете с толщиной слоя 30 мм.
В качестве раствора сравнения используют аликвотную часть анализируемого раствора без добавления молибденовокислого аммония.
Через все стадии анализа проводят контрольный опыт на содержание загрязнений в реактивах.
Результаты анализа вычисляют методом сравнения со стандартным образцом, близким по составу и массовой доле фосфора к анализируемой пробе и проведенным через все стадии анализа.
5.4. Обработка результатов
5.4.1. Массовую долю фосфора (X) в процентах по методу сравнения вычисляют по формуле
![]()
где C - массовая доля фосфора в стандартном образце, г;
Д1 - оптическая плотность анализируемого раствора;
Д2 - оптическая плотность стандартного раствора;
m - масса навески, г.
5.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 4).
Таблица 4
|
Абсолютные допускаемые расхождения, % |
|
|
До 0,03 |
0,003 |
|
Св. 0,03 до 0,06 |
0,004 |
|
" 0,06 " 0,10 |
0,006 |
|
" 0,10 " 0,20 |
0,010 |
6. ЭКСТРАКЦИОННО-ФОТОМЕТРИЧЕСКИЙ МЕТОД
ОПРЕДЕЛЕНИЯ ФОСФОРА В ЧУГУНАХ,
НИЗКОЛЕГИРОВАННЫХ И ВЫСОКОЛЕГИРОВАННЫХ
СТАЛЯХ
6.1. Сущность метода указана в п. 5.1.
Метод основан на экстрагировании желтой формы фосфорномолибденовой гетерополикислоты изоамиловым спиртом с последующим восстановлением до молибденовой сини раствором хлористого олова непосредственно в экстракте. При использовании данного варианта не мешают мышьяк, кремний, хром, никель. Влияние ванадия устраняют введением в раствор насыщенного раствора соли Мора, восстанавливая его при этом до 4-х валентного состояния.
6.2. Аппаратура, реактивы и растворы
Фотоколориметр ФЭК-56 или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота азотная по ГОСТ 4461-77 и разбавленная 1:1.
Кислота соляная по ГОСТ 3118-77.
Смесь кислот; готовят следующим образом: 3 объема соляной кислоты, плотностью 1,19 г/см3 смешивают с 1 объемом азотной кислоты, плотностью 1,4 г/см3.
Калий марганцовокислый по ГОСТ 20490-75, 4 %-ный раствор.
Натрий азотистокислый по ГОСТ 4168-79, 10 %-ный раствор.
Аммоний молибденовокислый по ГОСТ 3765-78; готовят следующим образом: 7,5 г аммония молибденовокислого растворяют при нагревании в 75 см3 воды, охлаждают и добавляют 25 см3 азотной кислоты, плотностью 1,4 г/см3.
Олово двухлористое по ГОСТ 36-78; готовят следующим образом: 2,4 г двухлористого олова растворяют в 30 см3 соляной кислоты, плотностью 1,19 г/см3, нагретой до кипения. После растворения приливают 70 см3 воды.
Растворы молибденовокислого аммония и двухлористого олова должны быть свежеприготовленными.
Спирт изоамиловый по ТУ 6-09-839.
Соль Мора по ГОСТ 4208-72, насыщенный раствор.
6.3. Проведение анализа
Навеску стали массой 0,25 г помещают в коническую колбу, вместимостью 100 см3, приливают 20 см3 азотной кислоты, разбавленной 1:1, и растворяют при нагревании.
При массовой доли хрома свыше 1 %, навеску образца растворяют в 15 см3 смеси кислот. Раствор выпаривают до сиропообразного состояния, приливают 5 см3 азотной кислоты, плотностью 1,4 г/см3 и вновь выпаривают до сиропообразного состояния. При содержании вольфрама в образце, операцию выпаривания повторяют дважды. Остаток растворяют в 10 см3 азотной кислоты, плотностью 1,4 г/см3, нагревают до растворения солей, приливают 10 см3 воды и кипятят до удаления окислов азота.
В горячие растворы приливают небольшими порциями марганцовокислый калий для окисления фосфора и кипятят до выпадения бурого осадка двуокиси марганца. Осадок растворяют в растворе азотистокислого натрия, который прибавляют по каплям до полного просветления раствора и кипятят раствор до удаления окислов азота. После охлаждения переносят в мерную колбу вместимостью 50 см3, доводят до метки водой и перемешивают. Если раствор мутный, фильтруют часть раствора через фильтр «белая лента», первые порции фильтрата отбрасывают.
В делительную воронку вместимостью от 100 до 150 см3 последовательно вводят 2 см3 раствора молибденовокислого аммония, 2 см3 анализируемого раствора и хорошо встряхивают. Если в образце присутствует ванадий, вводят 3 капли насыщенного раствора соли Мора и перемешивают. Из бюретки приливают 3 см3 изоамилового спирта и перемешивают в течение одной минуты. Дают раствору отстояться (для разделения фаз), затем осторожно по стенкам приливают 2 см3 раствора хлористого олова, слегка перемешивают и после разделения фаз нижний водный слой отбрасывают. Верхний органический слой переносят в кювету с толщиной слоя 5 мм и измеряют оптическую плотность на фотоколориметре, со светофильтром, имеющим область пропускания в интервале длин волн от 650 до 700 нм.
В качестве раствора сравнения используют изоамиловый спирт.
Параллельно с анализом пробы проводят холостой опыт, в качестве которого используют навеску карбонильного железа, проведенную через все стадии анализа.
При обработке результатов величину оптической плотности холостого опыта вычитают из величины оптической плотности анализируемого образца.
6.4. Обработка результатов
6.4.1. Массовую долю фосфора (X) в процентах по методу сравнения вычисляют по формуле, приведенной в п. 5.4.1.
6.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 4).
7. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ
ФОСФОРА И КРЕМНИЯ В УГЛЕРОДИСТЫХ СТАЛЯХ И
ЧУГУНАХ
(из одной навески)
7.1. Сущность метода
Сущность метода состоит в том, что растворение навески проводят в условиях необходимых для определения фосфора и первоначально создают кислотность (pH £ 0,5 н по серной кислоте), необходимую для образования фосфорно-молибденового комплекса, который восстанавливают гидроксиламином. После фотометрирования фосфорно-молибденового комплекса, в аликвотной части раствора, его разрушают добавлением аммония надсернокислого, создают кислотность для образования только кремнемолибденового комплекса, который восстанавливают тиомочевиной, в качестве катализатора используют медь сернокислую.
7.2. Аппаратура, реактивы и растворы
Фотоколориметр ФЭК-56 или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота азотная по ГОСТ 4461-77, разбавленная 1:6.
Калий марганцовокислый по ГОСТ 20490-75, 4 %-ный раствор.
Аммиак водный по ГОСТ 3760-64, разбавленный 1:9.
Гидроксиламин сернокислый по ГОСТ 7298-65, 20 %-ный раствор.
Кислота серная по ГОСТ 4207-77.
Медь сернокислая по ГОСТ 4165-78.
Аммоний надсернокислый (персульфат) по ГОСТ 20478-75, 12 %-ный раствор.
Аммоний молибденовокислый по ГОСТ 3765-78, 5 %-ный раствор.
Тиомочевина по ГОСТ 6344-73, 7 %-ный раствор.
Нейтрализованный раствор гидроксиламина; готовят следующим образом: 1000 см3 аммиака 1:9 смешивают с 1000 см3 20 %-ного раствора гидроксиламина сернокислого.
Аммоний молибденовокислый по ГОСТ 3765-78, раствор готовят следующим образом: к 2600 см3 воды приливают 400 см3 серной кислоты, плотностью 1,84 г/см3, затем не охлаждая жидкости растворяют 90 г аммония молибденовокислого.
Смесь меди сернокислой и серной кислоты, готовят следующим образом: к 600 см3 воды приливают 235 см3 серной кислоты, плотностью 1,84 г/см3 и 6,7 г меди сернокислой и доводят объем до 1 л водой.
7.3. Проведение анализа фосфора
Навеску стали или чугуна массой 0,1 г помещают в коническую колбу емкостью 100 см3 и растворяют в 5 см3 азотной кислоты, разбавленной 1:6, при умеренном нагревании. После растворения навески прибавляют 1 см3 калия марганцовокислого, кипятят 1 минуту, доводят объем до 25 см3 водой и прибавляют 4 см3 раствора гидроксиламина до полного просветления раствора, кипятят 1 минуту, охлаждают и количественно переводят в мерную колбу емкостью 50 см3. При анализе чугунов раствор переводят в мерную колбу через фильтр. Добавляют из бюретки по каплям 3 см3 раствора аммония молибденовокислого при энергичном встряхивании или пипеткой по стеночке колбы и затем хорошо перемешивают. Раствор выдерживают 15 - 20 минут до полного восстановления фосфорномолибденового комплекса и доводят до метки водой.
Оптическую плотность раствора измеряют на спектрофотометре при длине волны 670 нм или на фотоколориметре со светофильтром, имеющим область пропускания в интервале от 650 до 700 нм, в кювете с толщиной слоя 50 мм (для стали) или 30 мм (для чугуна) против воды.
Результаты анализа вычисляют методом сравнения со стандартным образцом, близким по составу и массовой доле фосфора с анализируемой пробой и проведенным через все стадии анализа.
7.4. Обработка результатов
7.4.1. Массовую долю фосфора (X) в процентах по методу сравнения вычисляют по формуле, приведенной в п. 5.4.1.
7.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 4) п. 5.4.2.
7.5. Проведение анализа кремния
Из мерной колбы емкостью 50 см3, после определения фосфора, отбирают аликвотную часть 5 см3 (в случае чугунов 2 см3 аликвотная часть и 6 см3 воды), помещают в мерную колбу, емкостью 100 см3 добавляют 10 см3 воды и 2 - 3 капли персульфата до полного осветления раствора, прибавляют 5 см3 5 %-ного раствора молибденовокислого аммония, выдерживают 5 мин. Прибавляют 15 см3 смеси меди сернокислой с серной кислотой и 15 см3 раствора тиомочевины, доводят водой до метки, перемешивают, выдерживают 15 минут. Измеряют оптическую плотность на спектрофотометре при длине волны 670 нм или на фотоколориметре со светофильтром, имеющим область пропускания в интервале от 650 до 700 нм, в кювете с толщиной слоя 10 мм.
Раствором сравнения служат все реактивы, проведенные через все стадии анализа.
Результаты анализа вычисляют методом сравнения со стандартным образцом, близким по составу и массовой доле кремния с анализируемой пробой и проведенным через все стадии анализа.
7.6. Обработка результатов
7.6.1. Массовую долю кремния (X) в процентах по методу сравнения вычисляют по формуле, приведенной в п. 8.4.2.
7.6.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 5), п. 8.4.3.
8. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ КРЕМНИЯ В СТАЛЯХ
Метод основан на образовании окрашенного в синий цвет комплексного соединения кремния с молибденовокислым аммонием в сернокислой среде с применением в качестве восстановителя двойной сернокислой соли закиси железа - аммония (соли Мора). Интенсивность окрашивания пропорциональна содержанию кремния. Чувствительность метода 0,000005 г в 100 см3 раствора. Кремнемолибденовая кислота устойчива в широком интервале кислотности вплоть до 5 н. Фосфор, присутствующий в растворе в виде фосфорной кислоты дает аналогичное окрашенное соединение с молибдатом аммония, влияние которого устраняется увеличением кислотности.
Важным моментом при определении кремния является растворение навески стали. При длительном растворении часть кремниевой кислоты может выделиться в виде геля и таким образом оказаться вне сферы реакции, образования комплексного соединения с молибдатом аммония.
В высоколегированных сталях для полного переведения кремния в растворимое состояние проводят щелочную обработку.
8.2. Аппаратура, реактивы и растворы
Спектрофотометр или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота соляная по ГОСТ 3118-77, плотностью 1,11 г/см3; готовят следующим образом: 150 мл соляной кислоты плотностью 1,19 г/см3 смешивают с 200 см3 воды.
Кислота азотная по ГОСТ 4461-77, плотностью 1,2 г/см3, готовят следующим образом: 240 см3 азотной кислоты плотностью 1,4 г/см3 смешивают с 340 см3 воды.
Кислота серная по ГОСТ 4204-77, разбавленная 1:3 и 1 %-ный раствор.
Аммоний молибденовокислый по ГОСТ 3765-78, 5 %-ный раствор, свежеприготовленный.
Соль Мора по ГОСТ 4208-72, 4 %-ный раствор; готовят следующим образом: 40 г соли Мора растворяют при перемешивании в 500 см3 воды, содержащей 50 см3 серной кислоты плотностью 1,82 г/см3, доводят объем до 1 л и перемешивают. Если раствор мутный, его отфильтровывают.
Стандартный раствор кремния А; готовят следующим образом; навеску массой 0,214 г двуокиси кремния по ГОСТ 9428-73, прокаленную при 1000 °С до постоянной массы, помещают в платиновую чашку, приливают 10 см3 20 %-ного раствора едкого натра и растворяют при нагревании. При отсутствии платины, растворение можно проводить в полиэтиленовом стакане на водяной бане. Охлажденный раствор переводят в мерную колбу вместимостью 1 л, доводят до метки водой и перемешивают.
1 см3 раствора содержит 0,0001 г кремния.
Раствор Б; готовят следующим образом: 20 см3 раствора А помещают в мерную колбу вместимостью 50 см3, доводят до метки водой и перемешивают. 1 см3 содержит 0,00004 г кремния. Разбавленный раствор готовят в день применения.
Раствор А хранят в полиэтиленовой посуде.
Установка титра стандартного раствора кремния.
50 см3 раствора А и 30 см3 соляной кислоты, разбавленной 1:4 выпаривают до состояния влажных солей, затем охлаждают до 50 °C добавляют 10 см3 соляной кислоты и 1, 2 см3 0,5 %-ного раствора желатины. Через 1 мин добавляют еще 2 см3 желатины и перемешивают. В течение 10 мин раствор отстаивается. Затем в него добавляют 50 см3 горячей воды, перемешивают, через 10 мин добавляют еще 50 см3 горячей воды и снова перемешивают, после чего фильтруют через фильтр с бумажной массой, промывают от 12 до 15 раз соляной кислотой, разбавленной 1:50, и прокаливают при температуре от 1000 до 1100 °C в платиновом тигле в течение 30 мин.
Тигель с осадком охлаждают в эксикаторе и взвешивают. Затем осадок осторожно смачивают от 3 до 5 каплями воды, приливают ее по стенкам тигля, прибавляют от 2 до 3 капель серной кислоты и от 3 до 5 см3 фтористоводородной кислоты.
Содержимое тигля выпаривают до прекращения выделения белых паров серной кислоты.
Тигель прокаливают в течение 15; 20 мин в муфельной печи при 1000 °C, охлаждают в эксикаторе и взвешивают. Параллельно проводят контрольный опыт на содержание загрязнений в реактивах.
Титр раствора (Т), выраженный в граммах кремния, вычисляют по формуле
![]()
где m1, m2 - масса тигля с осадком анализируемой пробы до и после обработки фтористоводородной кислотой соответственно, г;
m3, m4 - масса тигля с осадком контрольного опыта до и после обработки фтористоводородной кислотой соответственно, г;
0,4675 - коэффициент пересчета двуокиси кремния на кремний;
V - объем раствора кремния, взятый для установки титра, см3.
Навеску стали массой 0,2 г (при массовой доле кремния до 0,1 %) и 0,1 г (при массовой доле кремния свыше 0,1 %) помещают в коническую колбу или стакан вместимостью 100 см3, приливают 15 см3 соляной и 5 см3 азотной кислот и растворяют при нагревании, не доводя до кипения.
При анализе высоколегированных сталей по окончании растворения содержимое стакана количественно переносят в платиновую чашку, в которую предварительно помещают 20 см3 20 %-ного раствора едкого натра, отмеренного полиэтиленовой мензуркой. Щелочную жидкость нагревают до кипения для полного перехода кремния в растворимое состояние, нейтрализуют избыток щелочи соляной кислотой, разбавленной 1:1 до растворения гидроокисей и от 0,5 до 1,0 см3 в избытке и переводят в стакан, где проводилось растворение.
За отсутствием платины, переведение кремния в щелочную среду можно проводить в полиэтиленовых стаканах, нагревая их на водяной бане.
Охлажденный раствор переводят в мерную колбу, вместимостью 100 см3, доводят до метки водой и перемешивают.
Отбирают аликвотную часть 10 см3 (при массовой доле кремния до 0,1 %) и 5 см3 (при массовой доле кремния свыше 0,1 %), помещают в мерную колбу вместимостью 100 см3, приливают 20 см3 1 %-ного раствора серной кислоты, 10 см3 раствора молибденовокислого аммония и выдерживают 5 мин для развития желтого кремнемолибденового комплекса, прибавляют 20 см3 серной кислоты, разбавленной 1:3 для разрушения фосфорномолибденового комплекса и через 2 - 3 мин доводят раствором соли Мора до метки и перемешивают.
Оптическую плотность измеряют на спектрофотометре при длине волны 670 нм или на фотоколориметре со светофильтром, имеющим область пропускания в интервале длин волн от 650 до 700 нм, в кювете с толщиной слоя 30 или 50 мм.
В качестве раствора сравнения применяют навеску карбонильного железа, проведенную через все стадии анализа.
Массовую долю кремния определяют по градуировочному графику или методом сравнения со стандартным образцом, близким по составу к анализируемой пробе и проведенным через все стадии анализа.
8.3.1. Построение градуировочного графика для массовой доли кремния от 0,02 до 0,1 %,
В шесть стаканов вместимостью 100 см3 помещают навеску карбонильного железа, равную анализируемой пробе и 1,0; 2,0; 3,0; 4,0; 5,0 см3 стандартного раствора кремния Б, что соответствует 0,02; 0,04; 0,06; 0,08; 0,1 % кремния при навеске массой 0,2 г и аликвотной части раствора пробы 10 см3. Содержимое стакана растворяют в 15 см3 соляной кислоты плотностью 1,11 г/см3 и 5 см3 азотной кислоты плотностью 1,2 г/см3 при нагревании, не доводя до кипения. Далее анализ ведут как указано в п. 8.3.
Шестой стакан служит для приготовления раствора сравнения, который проводят через все стадии анализа.
Оптическую плотность измеряют в кюветах с толщиной слоя 50 мм.
По полученным значениям оптической плотности растворов строят градуировочный график.
8.4. Обработка результатов
8.4.1. Массовую долю кремния от 0,02 до 0,1 % (навеска массой 0,2 г, аликвотная часть раствора 10 см3) находят по градуировочному графику.
8.4.2. Массовую долю кремния (X) от 0,1 и выше в процентах по методу сравнения вычисляют по формуле
![]()
где C - массовая доля кремния в стандартном образце, г;
Д1 - оптическая плотность анализируемого раствора;
Д2 - оптическая плотность стандартного раствора;
m - масса навески, г.
8.4.3. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 5).
Таблица 5
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,01 до 0,02 |
0,006 |
|
Св. 0,02 " 0,05 |
0,010 |
|
" 0,05 " 0,10 |
0,015 |
|
" 0,10 " 0,25 |
0,02 |
|
" 0,25 " 0,50 |
0,03 |
|
" 0,50 " 1,00 |
0,04 |
|
" 1,00 " 2,00 |
0,07 |
|
" 2,0 " 4,00 |
0,10 |
9. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ КРЕМНИЯ В ЧУГУНАХ
9.1. Сущность метода указана в п. 8.1.
9.2. Аппаратура, реактивы и растворы
Спектрофотометр или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота серная по ГОСТ 4204-77, разбавленная 1:3 и 5 %-ная.
Калий марганцовокислый по ГОСТ 20490-75, 2 %-ный раствор.
Пергидроль по ГОСТ 10929-76, 3 %-ный раствор.
Аммоний молибденовокислый по ГОСТ 3765-78, 5 %-ный раствор, свежеприготовленный.
Соль Мора по ГОСТ 4208-72, 4 %-ный раствор; готовят следующим образом: 40 г соли Мора растворяют при перемешивании в 500 см3 воды, содержащей 50 см3 серной кислоты с плотностью 1,82 г/см3 и доводят объем до 1 л. Если раствор мутный его отфильтровывают.
Стандартный раствор кремния, приготовление указано в п. 8.2.
1 см3 раствора содержит 0,0001 г кремния.
9.3. Проведение анализа
Навеску чугуна массой 0,1 г помещают в коническую колбу вместимостью 250 см3, приливают 60 см3 5 %-ного раствора серной кислоты и растворяют при слабом нагревании. По окончании растворения в горячий раствор добавляют марганцовокислый калий до образования перекиси марганца (бурого цвета), которую разрушают 3 %-ным раствором перекиси водорода, добавляя ее по каплям.
Охлажденный раствор переводят в мерную колбу, вместимостью 200 см3, доводят до метки водой, перемешивают и часть раствора отфильтровывают в сухой стакан или колбу. Аликвотную часть 10 см3 помещают в мерную колбу, вместимостью 100 см3, приливают 10 см3 раствора молибденовокислого аммония и выдерживают в течении 5 мин для образования комплекса, прибавляют 20 см3 серной кислоты, разбавленной 1:3 и через 2 минуты доводят раствором соли Мора до метки и перемешивают.
Оптическую плотность измеряют на фотоколориметре со светофильтром, имеющим область пропускания в интервале длин волн от 650 до 700 нм или на спектрофотометре при длине волны 670 нм в кювете с толщиной слоя 10 мм.
В качестве раствора сравнения применяют навеску карбонильного железа, проведенную через все стадии анализа.
9.4. Обработка результатов
9.4.1. Массовую долю кремния (X) в процентах по методу сравнения вычисляют по формуле, приведенной в п. 8.4.2.
9.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 5).
10. ОБЪЕМНЫЙ ПЕРСУЛЬФАТНО-СЕРЕБРЯНЫЙ МЕТОД ОПРЕДЕЛЕНИЯ МАРГАНЦА В УГЛЕРОДИСТЫХ И НИЗКОЛЕГИРОВАННЫХ СТАЛЯХ, СОДЕРЖАЩИХ МЕНЕЕ 2,0 % ХРОМА
10.1. Сущность метода
Метод основан на окислении двухвалентного марганца в сернокислом растворе до семивалентного надсернокислым аммонием в присутствии азотнокислого серебра. Образовавшаяся марганцовая кислота, окрашивающая раствор в характерно фиолетово-красный цвет, восстанавливается серноватистокислым натрием. Определению марганца мешают наличие хрома свыше 1,5 % и ионы хлора.
Кислота серная по ГОСТ 4204-77.
Кислота азотная по ГОСТ 4461-77.
Кислота ортофосфорная по ГОСТ 6552-80.
Серебро азотнокислое по ГОСТ 1277-75, 0,5 %-ный раствор.
Смесь кислот для растворения; готовят следующим образом: к 845 см3 воды приливают при перемешивании 120 см3 серной кислоты, 15 см3 азотной кислоты, 20 см3 ортофосфорной кислоты и 2,5 см3 раствора азотнокислого серебра.
Аммоний надсернокислый (персульфат) по ГОСТ 20478-75, 12 %-ный раствор, свежеприготовленный.
Натрий серноватистокислый (тиосульфат), титрованный раствор; готовят следующим образом: 0,6 г серноватистокислого натрия и 0,2 г азотистокислого натрия растворяют в 100 см3 воды, переводят в мерную колбу вместимостью 1 л и перемешивают. Титр раствора тиосульфата натрия устанавливают по стандартному образцу, близкому по химическому составу и содержанию марганца к анализируемой пробе и проведенному через все стадии анализа.
Титр раствора тиосульфата натрия (Т), выраженный в граммах, вычисляют по формуле:
![]()
где C - массовая доля марганца в стандартном образце, %;
m - масса навески стандартного образца, г;
V - объем раствора тиосульфата натрия, израсходованный на титрование, см3.
10.3. Проведение анализа
Навеску стали или чугуна массой 0,5 г (при массовой доле марганца от 0,1 до 0,5 %), 0,2 г (при массовой доле марганца свыше 0,5 % до 1,0 %) и 0,1 г (при массовой доле марганца свыше 1,0 %) помещают в коническую колбу вместимостью 250 см3 и растворяют в 40 см3 смеси при умеренном нагревании. По окончании растворения кипятят мин 2 - 3 для удаления окислов азота. При анализе чугуна осадок графита и кремниевой кислоты отфильтровывают и промывают от 5 до 6 раз горячей водой, разбавляют водой до 100 - 130 см3, приливают 5 см3 раствора азотнокислого серебра, 30 см3 раствора персульфата аммония, нагревают до кипения и выдерживают в теплом месте до прекращения выделения пузырьков кислорода. Раствор охлаждают в проточной воде до комнатной температуры (от 20 до 22 °C) и немедленно титруют раствором тиосульфата натрия, приливая его с постоянной скоростью до перехода окраски титруемого раствора в слаборозовую. После этого раствор тиосульфата натрия прибавляют по каплям до полного исчезновения розовой окраски.
10.4. Обработка результатов
10.4.1. Массовую долю марганца (X) в процентах вычисляют по формуле
![]()
где Т - титр раствора тиосульфата натрия, выраженный в г марганца;
V - объем раствора тиосульфата натрия, израсходованный на титрование, см3;
m - масса навески, г.
10.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 6).
Таблица 6
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,1 до 0,20 |
0,010 |
|
Св. 0,2 " 0,40 |
0,020 |
|
" 0,40 " 1,00 |
0,030 |
|
0,040 |
|
|
" 2,00 " 3,00 |
0,050 |
|
" 3,00 " 5,00 |
0,08 |
|
" 5,00 |
0,12 |
11. ОБЪЕМНЫЙ ПЕРСУЛЬФАТНО-СЕРЕБРЯНЫЙ МЕТОД ОПРЕДЕЛЕНИЯ МАРГАНЦА В ВЫСОКОЛЕГИРОВАННЫХ СТАЛЯХ
11.1. Сущность метода указана в п. 9.1.
Определению марганца мешает присутствие хрома (при массовой доле его больше 1,5 %), так как в условиях анализа хром окисляется в хромовую кислоту желтого цвета, которая сильно затрудняет титрование.
Отделение марганца от большинства элементов, в том числе и от хрома, достигается осаждением их водной суспензией окиси цинка.
11.2. Реактивы и растворы
Кислота серная по ГОСТ 4204-77, разбавленная 1:4.
Кислота азотная по ГОСТ 4461-77.
Кислота азотная по ГОСТ 4461-77.
Кислота соляная по ГОСТ 3118-77, разбавленная 1:1.
Аммиак водный по ГОСТ 3760-79.
Окись цинка по ГОСТ 10262-73; готовят следующим образом: г 20 - 25 окиси цинка помещают в стакан и приливают см3 150 - 200 воды. Перед употреблением смесь хорошо размешивают стеклянной палочкой для придания ей однородной консистенции.
Смесь кислот; приготовление указано в п. 10.2.
Серебро азотнокислое, 0,5 %-ный раствор. Хранят в склянке из темного стекла.
Надсернокислый аммоний (персульфат), 20 %-ный раствор.
Титрованный раствор серноватистокислого натрия (тиосульфата); приготовление и установка титра указаны в п. 10.2.
11.3. Проведение анализа
Навеску стали массой 1,0 г (при массовой доле марганца до 0,5 %), 0,5 г (при массовой доле марганца свыше 0,5 % до 5,0 %) и 0,25 г (при массовой доле марганца свыше 5,0 %) помещают в коническую колбу вместимостью 250 см3, приливают 40 см3 серной кислоты, разбавленной 1:4 и растворяют при нагревании. По окончании растворения прибавляют по каплям азотную кислоту до прекращения вспенивания и кипятят до удаления окислов азота.
Если сталь не растворяется в серной кислоте, то растворение ведут в царской водке или в соляной кислоте, разбавленной 1:1 до полного растворения навески и разложения карбидов, после чего приливают 10 см3 серной кислоты плотностью 1,82 г/см3 и дважды выпаривают раствор до появления паров серной кислоты.
После растворения навески и полного удаления ионов хлора, содержимое колбы переводят количественно в мерную колбу вместимостью 250 см3, осторожно по каплям добавляют аммиак водный для нейтрализации избытка кислоты, но не до выпадения гидроокисей и небольшими порциями приливают суспензию окиси цинка до появления ее нерастворяющегося избытка на дне колбы в виде осадка. Содержимое колбы разбавляют водой (но не до метки), хорошо перемешивают, охлаждают, затем доводят до метки водой и вновь перемешивают. После отстаивания и просветления часть раствора отфильтровывают через фильтр «красная лента», первые порции фильтрата отбрасывают.
Отбирают аликвотную часть раствора 50 см3 (при массовой доле марганца до 5 %) и 25 см3 (при массовой доле марганца свыше 5 %), помещают в коническую колбу вместимостью 250 см3, приливают 25 см3 смеси кислот и кипятят. В горячий раствор приливают 5 см3 раствора, азотнокислого серебра, от 25 до 30 см3 раствора персульфата аммония, нагревают до кипения и выдерживают в теплом месте до прекращения выделения пузырьков кислорода. Раствор охлаждают в проточной воде до комнатной температуры (от 20 до 22 °C) и немедленно титруют раствором серноватистокислого натрия, приливая с постоянной скоростью до перехода окраски титруемого раствора в слаборозовую. После этого титрованный раствор прибавляют по каплям до полного исчезновения розовой окраски.
11.4.1. Массовую долю марганца (X) в процентах вычисляют по формуле, приведенной в п. 10.4.1.
11.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений указаны в (табл. 6), п. 10.4.2.
12. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ МАРГАНЦА В ВЫСОКОЛЕГИРОВАННЫХ СТАЛЯХ
12.1. Сущность метода
Метод основан на окислении двухвалентного марганца в марганцовую кислоту в кислой среде персульфатом аммония в присутствии катализатора - азотнокислого серебра. Образующаяся марганцовая кислота, окрашивающая раствор в характерный малиново-красный цвет, имеет максимум светопоглащения при λ = 525 нм. Интенсивность окраски пропорциональна массовой доле марганца. Чувствительность метода 0,000016 г в 100 см3 раствора.
При наличии в растворе ионов хлора, их предварительно удаляют выпариванием с серной кислотой. Для устранения возможного образования четырехвалентного марганца, вводят фосфат ион в виде ортофосфорной кислоты, который стабилизирует промежуточную форму трехвалентного марганца и связывает трехвалентное железо в бесцветный комплекс.
Определению мешают значительные количества окрашенных ионов хрома, никеля, кобальта, меди, церия четырехвалентного. При высоком содержании указанных элементов полной компенсации их окраски удается достигнуть путем использования части анализируемого раствора в качестве раствора сравнения, введя в него селенистую кислоту или азотистокислый натрий. При этом окраска марганцовой кислоты разрушается. Такой способ компенсирующего влияния дает хорошие результаты даже при количествах присутствующих элементов в 200 - 300 раз превышающих содержание марганца.
12.2. Аппаратура, реактивы и растворы
Фотоколориметр ФЭК-56 или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота серная по ГОСТ 4204-77, разбавленная 1:4.
Кислота ортофосфорная по ГОСТ 6552-80, разбавленная 1:1.
Кислота азотная по ГОСТ 4461-77.
Кислота соляная по ГОСТ 3118-77.
Серебро азотнокислое по ГОСТ 1277-75, 0,25 %-ный раствор.
Аммоний надсернокислый (персульфат) по ГОСТ 20478-75, 30 %-ный раствор.
Натрий азотистокислый по ГОСТ 4168-79, 2 %-ный раствор.
Кислота селенистая ч.д.а., 2 %-ный раствор.
12.3. Проведение анализа
Навеску массой 0,5 г (при массовой доле марганца до 0,8 %) и 0,25 г (при массовой доле марганца свыше 0,8 %) помещают в коническую колбу вместимостью 250 см3, приливают 40 см3 серной кислоты разбавленной 1:4 и растворяют при нагревании. По окончании растворения прибавляют по каплям азотную кислоту до прекращения вспенивания и нагревают до удаления окислов азота.
При анализе сложных сталей, растворение навески производят в 20 см3 смеси соляной и азотной кислот при соотношении 1:1. По окончании растворения приливают 10 см3 серной кислоты, разбавленной 1:1 и упаривают до паров серной кислоты для освобождения от ионов хлора. Выпавшие соли растворяют при нагревании в 20 см3 воды.
Охлажденный раствор переводят в мерную колбу вместимостью 100 см3, доводят до метки водой и перемешивают.
Аликвотную часть 20 см3 помещают в коническую колбу 100 см3, приливают 5 см3 ортофосфорной кислоты, слегка взбалтывают и добавляют последовательно 10 см3 раствора азотнокислого серебра и 15 см3 раствора надсернокислого аммония. Раствор нагревают до полного окисления марганца (но не более 30 - 40 с), дают постоять в теплом месте для полного разрушения надсернокислого аммония, охлаждают, переводят в мерную колбу вместимостью 100 см3, доводят до метки водой и перемешивают.
Оптическую плотность измеряют на фотоколориметре со светофильтром, имеющим область пропускания в интервале длин волн от 510 до 550 нм или на спектрофотометре при длине волны 525 нм в кювете с толщиной слоя 20 - 50 мм относительно раствора сравнения, которым служит оставшийся раствор пробы, налитый в другую кювету, с добавлением в него от 2 до 3 капель селенистой кислоты или раствора азотистокислого натрия, разрушающих окраску марганцовой кислоты.
12.4. Обработка результатов
12.4.1. Массовую долю марганца (X) в процентах по методу сравнения вычисляют по формуле
![]()
где C - массовая доля марганца в стандартном образце или стандартном растворе, г;
Д1 - оптическая плотность анализируемого раствора;
Д2 - оптическая плотность стандартного раствора;
m - масса навески, г.
12.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений указанных (в табл. 6).
13. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ ХРОМА (0,01 - 0,05 %)
13.1. Сущность метода
Метод основан на окислении дифенилкарбазида шестивалентным хромом в сернокислой среде (от 0,2 до 0,5 н) и измерении интенсивности окраски полученного соединения, окрашенного в краснофиолетовый цвет (λ = 536 нм). Влияние трехвалентного железа устраняют прибавлением фосфорной кислоты.
13.2. Аппаратура, реактивы и растворы
Фотоколориметр ФЭК-56 или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота серная по ГОСТ 4204-77.
Кислота ортофосфорная по ГОСТ 6552-58.
Смесь кислот; готовят следующим образом: к 700 см3 воды осторожно при непрерывном перемешивании, приливают 150 см3 серной кислоты, охлаждают, приливают 150 см3 ортофосфорной кислоты.
Кислота азотная по ГОСТ 4461-77.
Серебро азотнокислое по ГОСТ 1277-75, 0,5 %-ный раствор (хранить в посуде из темного стекла).
Аммоний надсернокислый (персульфат аммония) по ГОСТ 204-75, 10 %-ный свежеприготовленный раствор.
Дифенилкарбазид по ГОСТ 5859-70, 0,1 %-ный свежеприготовленный раствор; готовят следующим образом: 0,1 г реактива растворяют в 10 см3 этилового спирта и доливают до 100 см3 водой.
Спирт этиловый ректификованный по ГОСТ 5962-67.
13.3. Проведение анализа
Навеску стали или чугуна массой 0,2 г (при массовой доле хрома от 0,01 до 0,10 %) или 0,1 г (при массовой доле хрома свыше 0,1 %) помещают в коническую колбу вместимостью 250 см3, приливают 20 см3 смеси кислот, накрывают часовым стеклом и растворяют пробу при умеренном нагревании. Затем снимают стекло и обмывают его над колбой небольшим количеством воды, приливают азотную кислоту по каплям до прекращения вспенивания раствора и кипятят этот раствор до удаления окислов азота. При наличии осадка раствор отфильтровывают через ватный тампон, промывают от 5 до 6 раз горячей водой, собирая фильтрат и промывные воды в колбу вместимостью 100 см3, затем прибавляют 1 мл раствора азотнокислого серебра и 10 см3 раствора надсернокислого аммония. Полученный раствор постепенно нагревают до появления розовой окраски, после чего кипятят от 2 до 3 мин до полного разрушения избытка персульфата.
Раствор охлаждают, переводят в мерную колбу вместимостью 100 см3 доливают до метки водой и перемешивают.
В мерную колбу вместимостью 50 см3 помещают аликвотную часть раствора 10 см3, приливают 5 см3 раствора дифенилкарбазида и перемешивают, через 10 мин доливают раствор до метки водой, снова перемешивают и через 5 минут измеряют оптическую плотность растворов на спектрофотометре при длине волны 536 нм или на фотоколориметре со светофильтром, имеющим область пропускания в интервале длин волн от 530 до 550 нм в кювете с толщиной слоя 50 мм.
Раствор для сравнения готовят следующим образом: в мерную колбу вместимостью 50 см3 приливают 10 см3 воды, 2 см3 смеси кислот, 5 см3 раствора дифенилкарбазида, доливают до метки водой и перемешивают.
13.4. Обработка результатов
13.4.1. Массовую долю хрома (X) в процентах по методу сравнения вычисляют по формуле
![]()
где C - массовая доля хрома в стандартном образце, г;
Д1 - оптическая плотность анализируемого раствора;
Д2 - оптическая плотность стандартного раствора;
m - масса навески, г.
13.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных в (табл. 7).
Таблица 7
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,01 до 0,025 |
0,005 |
|
Св. 0,025 " 0,050 |
0,007 |
|
" 0,050 " 0,10 |
0,010 |
|
" 0,10 " 0,20 |
0,015 |
|
" 0,20 " 0,50 |
0,020 |
14. ОБЪЕМНЫЙ ПЕРСУЛЬФАТНО-СЕРЕБРЯНЫЙ МЕТОД ОПРЕДЕЛЕНИЯ ХРОМА (0,6 - 35 %)
14.1. Сущность метода
Метод основан на окислении трехвалентного хрома надсернокислым аммонием до шестивалентного в сернокислой среде (от 0,05 до 0,5 н) в присутствии азотнокислого серебра. Хромовую кислоту восстанавливают раствором соли двухвалентного железа, избыток которого оттитровывают раствором марганцовокислого калия.
14.2. Реактивы и растворы
Кислота серная по ГОСТ 4204-77 и разбавленная 5:95 и 1:1.
Кислота ортофосфорная по ГОСТ 6552-80.
Смесь кислот; готовят следующим образом: к 800 см3 воды осторожно при непрерывном перемешивании приливают 200 см3 серной кислоты, охлаждают, приливают 80 см3 ортофосфорной кислоты и перемешивают.
Кислота азотная по ГОСТ 4461-77.
Серебро азотнокислое по ГОСТ 1277-75, 0,5 %-ный раствор (хранят в посуде из темного стекла).
Аммоний надсернокислый (персульфат) по ГОСТ 20478-75, 20 %-ный свежеприготовленный раствор.
Натрий хлористый по ГОСТ 4233-77, 5 %-ный раствор.
Двойная сернокислая соль закиси железа и аммония (соль Мора) по ГОСТ 4208-72, титрованный раствор; готовят следующим образом: 40 г соли растворяют в 400 - 500 см3 серной кислоты, разбавленной 1:95, раствор переносят в мерную колбу вместимостью 1 л, доливают серной кислотой, разбавленной 1:95, до метки и перемешивают.
Калий марганцовокислый по ГОСТ 20490-75, титрованный раствор; готовят следующим образом: 3,16 г марганцовокислого калия растворяют в 500 см3 воды, раствор переносят в мерную колбу вместимостью 1 л, доливают до метки водой, перемешивают и выдерживают от 7 до 10 суток. Раствор хранят в посуде из темного стекла.
Титр раствора устанавливают по безводному щавелевокислому натрию и проверяют по стандартному образцу стали или чугуна.
Для установки титра раствора марганцовокислого калия, 0,1 г безводного щавелевокислого натрия по ГОСТ 5839-77, растворяют при нагревании в 50 см3 воды, добавляют 15 см3 серной кислоты, разбавленной 1:1, нагревают до температуры от 70 до 80 °C и титруют раствором марганцовокислого калия до появления устойчивой розовой окраски. Для вычисления титра берут среднее арифметическое от 3 до 4 результатов.
Титр раствора марганцовокислого калия (Т) выраженный в граммах хрома, вычисляют по формуле
![]()
где 0,259 - коэффициент пересчета титра раствора марганцовокислого калия, установленного по щавелевокислому натрию, на титр, выраженный в граммах хрома;
m - масса навески щавелевокислого натрия, г;
V - объем раствора марганцовокислого калия, израсходованный на титрование, см3.
Титр раствора проверяют по трем навескам стандартного образца стали или чугуна с массовой долей хрома и других элементов, близким к массовой доле их в испытуемом образце.
Соотношение объемов растворов соли Мора и марганцовокислого калия (K) устанавливают следующим образом: в коническую колбу вместимостью 250 см3 наливают из бюретки 25 см3 раствора соли Мора и титруют раствором марганцовокислого калия до появления устойчивой (в течение от 2 до 3 мин) слаборозовой окраски.
Соотношение объемов растворов (K) вычисляют по формуле
![]()
где V1 - объем раствора марганцовокислого калия, израсходованный на титрование 25 см3 раствора соли Мора, см3;
V - объем раствора соли Мора, взятый для установления соотношения, см3 (25 см3).
Соотношение устанавливают три раза и берут среднее значение. Соотношение следует проверять ежедневно.
14.3. Проведение анализа
Навеску стали массой 1,0 г (при массовой доле хрома до 2,0 %) 0,5 г (при массовой доле хрома свыше 2,0 до 5,0 %), 0,20 г (при массовой доле хрома свыше 5,0 до 10,0 %) и 0,1 г (при массовой доле хрома свыше 10,0 до 30 %) помещают в коническую колбу, вместимостью 500 см3 и растворяют при умеренном нагревании в 60 см3 смеси кислот. После полного растворения прибавляют по каплям азотную кислоту до прекращения вспенивания и кипятят до полного разрушения карбидов и удаления карбидов и удаления окислов азота. Если сталь не растворяется в смеси серной и ортофосфорной кислот, навеску растворяют в 30 см3 соляной и 10 см3 азотной кислот. После чего к раствору доливают смесь кислот (при массовой доле вольфрама свыше 5 % вливают еще от 5 до 10 см3 ортофосфорной кислоты) и раствор выпаривают до появления паров серной кислоты. Содержимое колбы охлаждают, приливают от 150 до 200 см3 воды и нагревают до растворения солей. При анализе сталей c массовой долей марганца менее 0,1 % в раствор вводят 1 см3 0,5 %-ного раствора сернокислого марганца. К полученному раствору приливают 10 см3 раствора азотнокислого серебра, от 20 до 40 см3 раствора надсернокислого аммония и нагревают до кипения. Момент полного окисления хрома определяют по появлению малиновой окраски образующейся марганцевой кислоты.
Раствор кипятят до полного разрушения персульфата аммония. К кипящему раствору приливают 5 см3 раствора хлористого натрия и продолжают кипячение до исчезновения малиновой окраски. Цвет раствора должен стать желтым. Если малиновая окраска раствора долго не исчезает или осадок хлористого серебра получается бурого цвета, необходимо добавить еще от 1 до 2 см3 раствора хлористого натрия и продолжать кипячение до исчезновения малиновой окраски. Раствор охлаждают в проточной воде до комнатной, приливают из бюретки точно отмеренное количество 0,1 н раствора соли Мора до исчезновения желтой и появления зеленой окраски и сразу же оттитровывают избыток раствора соли Мора раствором марганцовокислого калия до появления устойчивой (в течение от 2 до 3 минут) розовой окраски.
14.4. Обработка результатов
14.4.1. Массовую долю хрома (X) в процентах вычисляют по формуле
![]()
где T - титр раствора марганцовокислого калия, выраженный в граммах хрома;
V - объем раствора соли Мора, взятый с избытком для восстановления хрома, см3;
V1 - объем раствора марганцовокислого калия, израсходованный на титрование избытка раствора соли Мора, см3;
m - масса навески пробы, г;
K - соотношение объемов раствора соли Мора и марганцовокислого калия.
14.4.2. Абсолютные допускаемые расхождения между результатами трех параллельных определений не должны превышать значений, указанных (в табл. 8).
Таблица 8
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,5 до 1,0 |
0,03 |
|
Св. 1,0 " 2,0 |
0,05 |
|
" 2,0 " 5,0 |
0,07 |
|
" 5,0 " 10,0 |
0,10 |
|
" 10,0 " 15,0 |
0,15 |
|
" 15,0 " 20,0 |
0,20 |
|
" 20,0 |
0,25 |
15. ОБЪЕМНЫЙ ПЕРСУЛЬФАТНО-СЕРЕБРЯНЫЙ МЕТОД
ОПРЕДЕЛЕНИЯ ХРОМА (0,6 - 35 %)
С ФЕНИЛАНТРАНИЛОВОЙ КИСЛОТОЙ В СТАЛЯХ,
НЕ СОДЕРЖАЩИХ ВАНАДИЙ
Метод основан на окислении трехвалентного хрома надсернокислым аммонием до шестивалентного в сернокислой среде от 0,05 до 0,5 н) в присутствии азотнокислого серебра. Хромовую кислоту восстанавливают раствором соли двухвалентного железа. В качестве окислительно-восстановительного индикатора применяют фенилантраниловую кислоту.
15.2. Реактивы и растворы
Кислота серная по ГОСТ 4204-77 и разбавленная 5:95 и 1:4.
Кислота ортофосфорная по ГОСТ 6552-80.
Кислота азотная по ГОСТ 4461-77.
Серебро азотнокислое по ГОСТ 1277-75, 0,5 %-ный раствор (хранят в склянке из темного стекла).
Аммоний надсернокислый (персульфат) по ГОСТ 20478-75, годный свежеприготовленный раствор.
Натрий хлористый по ГОСТ 4233-77, 5 %-ный раствор.
Двойная сернокислая соль закиси железа и аммония (соль Мора) по ГОСТ 4208-72, титрованный раствор; приготовление раствора описано в п. 13.2.
Фенилантраниловая кислота 0,1 %-ный раствор; готовят следующим образом: 0,1 г кислоты и 0,1 г безводного углекислого натрия растворяют при нагревании в 100 см3 воды.
15.3. Проведение анализа
Навеску стали массой 1 г (при массовой доле хрома от 0,5 до 2,00 %), 0,5 г (при массовой доле хрома от 2,0 до 5,00 %), 0,2 г (при массовой доле хрома от 5,00 до 10,0 %), 0,1 г (при массовой доле хрома свыше 10,00 %) помещают в коническую колбу вместимостью 500 см3 и растворяют при нагревании в 40 см3 серной кислоты, разбавленной 1:4 и 10 см3 ортофосфорной кислоты. После полного растворения прибавляют по каплям азотную кислоту до прекращения вспенивания и выпаривают дважды до паров серной кислоты.
Если сталь не растворяется в серной кислоте, то растворение проводят в соляной кислоте, разбавленной 1:1 или в царской водке с последующим добавлением серной и фосфорной кислот и упариванием растворов дважды до паров серной кислоты.
Соли растворяют в 200 см3 горячей воды, приливают 10 см3 раствора азотнокислого серебра, от 20 до 40 см3 раствора надсернокислого аммония и нагревают до кипения. Момент полного окисления хрома определяют по появлению малиновой окраски образующейся марганцовой кислоты. Раствор кипятят до полного разрушения персульфата аммония. К кипящему раствору приливают 5 см3 раствора хлористого натрия и продолжают кипячение до исчезновения малиновой окраски. Цвет раствора должен стать желтым. Если малиновая окраска раствора долго не исчезает или осадок хлористого серебра получается бурого цвета, необходимо добавить еще от 1 до 2 см3 раствора хлористого натрия и продолжать кипячение до исчезновения малиновой окраски. Раствор охлаждают в проточной воде до комнатной, приливают 10 см3 серной кислоты плотностью 1,82 г/см3, от 5 до 6 капель фенилантраниловой кислоты и титруют раствором соли Мора до перехода окраски из краснофиолетовой в зеленую.
15.4. Обработка результатов
15.4.1. Массовую долю хрома (X) в процентах по методу сравнения вычисляют по формуле
![]()
где C - массовая доля хрома в стандартном образце, г;
V1 - объем раствора соли Мора, израсходованный на титрование анализируемого образца, см3;
V2 - объем раствора соли Мора, израсходованный на титрование стандартного образца, см3;
m - масса навески, г.
15.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 8).
16. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ НИКЕЛЯ
(0,025 - 0,5 %) В УГЛЕРОДИСТЫХ И НИЗКОЛЕГИРОВАННЫХ СТАЛЯХ
С ДИМЕТИЛГЛИОКСИМОМ
Метод основан на образовании окрашенного в коричнево-красный цвет комплексного соединения никеля с диметилглиоксимом в щелочной среде. В качестве окислителя применяют надсернокислый аммоний; Интенсивность окраски пропорциональна массовой доле никеля и устойчива длительное время. Чувствительность метода 0,000005 г в 100 см3 раствора. Максимум светопоглощения находится при λ = 460 - 480 нм. Мешающее влияние железа, хрома и других элементов устраняют комплексованием их раствором калия-натрия виннокислого.
16.2. Аппаратура, реактивы и растворы
Фотоколориметр ФЭК-56 или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота азотная по ГОСТ 4461-77, разбавленная 1:3.
Калий-натрий виннокислый по ГОСТ 5845-79, 20 %-ный раствор.
Аммоний надсернокислый по ГОСТ 20478-75, 20 %-ный раствор, свежеприготовленный.
Натр едкий по ГОСТ 4328-77, 20 %-ный раствор.
Диметилглиоксим по ГОСТ 5828-77, 0,5 %-ный раствор в 20 %-ном растворе едкого натра.
16.3. Проведение анализа
Навеску массой 0,1 г помещают в коническую колбу или стакан вместимостью 100 см3, приливают 10 см3 азотной кислоты, разбавленной 1:3 и растворяют при умеренном нагревании. После растворения навески раствор кипятят до удаления бурых окислов азота. Раствор охлаждают, переводят в мерную колбу, вместимостью 100 см3, доводят до метки водой и перемешивают. Отбирают аликвотную часть 25 см3 в мерную колбу вместимостью 100 см3, приливают 10 см3 калия-натрия виннокислого, 5 см3 раствора едкого натра, 5 см3 раствора надсернокислого аммония, 10 см3 раствора диметилглиоксима, сразу доводят до метки водой и перемешивают. После добавления каждого реактива раствор тщательно перемешивают.
Раствор для сравнения готовят следующим образом: мерную колбу вместимостью 100 см3 помещают вторую аликвотную часть раствора 10 см3, приливают все реактивы в том же количестве за исключением раствора диметилглиоксима, доливают до метки водой и перемешивают.
Оптическую плотность измеряют на фотоколориметре со светофильтром, имеющим область пропускания в интервале от 460 до 480 нм или на спектрофотометре при длине волны 460 нм, в кювете с толщиной слоя 30 мм.
16.4. Обработка результатов
16.4.1. Массовую долю никеля (X) в процентах по методу сравнения вычисляют по формуле
![]()
где C - массовая доля никеля в стандартном образце или в стандартном растворе, г;
Д1 - оптическая плотность анализируемого раствора;
Д2 - оптическая плотность стандартного раствора;
m - масса навески, г.
16.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 9).
Таблица 9
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,025 до 0,04 |
0,008 |
|
Св. 0,04 " 0,08 |
0,010 |
|
" 0,08 " 0,15 |
0,015 |
|
" 0,15 " 0,30 |
0,020 |
|
" 0,30 " 0,50 |
0,030 |
|
" 0,50 " 1,00 |
0,04 |
|
" 1,00 " 2,00 |
0,05 |
|
" 2,0 " 5,00 |
0,07 |
|
" 5,00 " 10,0 |
0,09 |
17. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ НИКЕЛЯ (0,5 - 10 %) В ЛЕГИРОВАННЫХ СТАЛЯХ
17.1. Сущность метода указана в п. 16.1.
17.2. Аппаратура, реактивы и растворы
Фотоколориметр ФЭК-56 или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота азотная по ГОСТ 4461-77 и разбавленная 1:1.
Кислота соляная по ГОСТ 3118-77, разбавленная 1:1.
Калий натрий виннокислый (сегнетовая соль), 20 %-ный раствор, свежеприготовленный.
Аммоний надсернокислый по ГОСТ 20478-75, 20 %-ный раствор, свежеприготовленный.
Натр едкий по ГОСТ 4328-77, 20 %-ный раствор.
Диметилглиоксим по ГОСТ 5828-77, 0,5 %-ный раствор в 20 %-ном растворе едкого натра.
17.3. Проведение анализа
Навеску стали массой 0,1 г помещают в стакан вместимостью 100 см3, приливают 10 см3 соляной кислоты, разбавленной 1:1, 10 см3 азотной кислоты, разбавленной 1:1 и растворяют при нагревании. По окончании растворения, раствор охлаждают, переводят в мерную колбу вместимостью 100 см3, доводят до метки водой и перемешивают. Отбирают аликвотную часть 10 см3 (при массовой доле никеля до 5,0 %) и 5,0 см3 (при массовой доле никеля свыше 5,0 %), помещают в мерную колбу вместимостью 100 см3, приливают 10 см3 раствора калия-натрия виннокислого, 5 см3 раствора персульфата аммония, 5 см3 раствора едкого натра и 10 см3 раствора диметилглиоксима. После прибавления каждого реактива раствор в мерной колбе перемешивают. Через мин. 3 - 5 раствор разбавляют до метки водой, перемешивают и измеряют оптическую плотность на фотоколориметре со светофильтром, имеющим область пропускания в интервале длин волн от 460 до 480 нм или на спектрофотометре при длине волны 460 нм в кювете с толщиной слоя 5 мм.
В качестве раствора сравнения используют вторую аликвотную часть с добавлением всех реактивов в том же количестве без диметилглиоксима.
17.4. Обработка результатов
17.4.1. Массовую долю никеля (X) в процентах по методу сравнения вычисляют по формуле, указанной в п. 16.4.1.
17.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 9).
18. ДИФФЕРЕНЦИАЛЬНЫЙ СПЕКТРОФОТОМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ НИКЕЛЯ В ЛЕГИРОВАННЫХ СТАЛЯХ
18.1. Сущность метода указана в п. 15.1.
Метод дифференциональный спектрофотометрии основан на фотометрировании испытуемых растворов, концентрация светопоглощающего вещества в которых больше концентрации раствора сравнения.
Для повышения точности измерений следует стремиться к максимально высокой оптической плотности раствора сравнения и к максимальному сближению значений До и Дисп, т.е. концентрации испытуемого раствора и раствора сравнения должны быть максимально высокими (но измеримыми) и предельно близкими.
Процент ошибки зависит прежде всего от точности показания прибора, от характера калибровочного графика, от точности построения калибровочного графика и способа обработки результатов измерения.
18.2. Аппаратура, реактивы и растворы
Фотоколориметр ФЭК-56 или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота соляная по ГОСТ 3118-77, разбавленная 1:1.
Кислота азотная по ГОСТ 4461-77.
Калий-натрий виннокислый (сегнетовая соль) по ГОСТ 5845-70, 20 %-ный раствор, свежеприготовленный.
Аммоний надсернокислый (персульфат) по ГОСТ 204-78, 20 %-ный раствор, свежеприготовленный.
Натр едкий по ГОСТ 4328-77, 20 %-ный раствор.
Диметилглиоксим по ГОСТ 5828-77, 0,5 %-ный раствор в 20 %-ном растворе едкого натра.
Стандартный раствор никеля А; готовят следующим образом: 0,5 г металлического никеля растворяют при умеренном нагревании в 35 см3 азотной кислоты, разбавленной 3:2. Раствор выпаривает до 3 - 5 см3, приливают 40 см3 воды и нагревают до полного просветления раствора. Затем охлаждают, переводят в мерную колбу вместимостью 1 л, доливают до метки водой и перемешивают. 1 см3 раствора содержит 0,0005 г никеля.
Раствор Б; готовят следующим образом: 25 см3 раствора А помещают в мерную колбу вместимостью 250 см3, разбавляют до метки водой и перемешивают, 1 см3 раствора Б содержит 0,00005 г никеля.
Навеску стали массой 0,1 г помещают в стакан вместимостью 100 см3, приливают 20 см3 соляной кислоты разбавленной 1:1 и растворяют при нагревании. По окончании растворения прибавляют 5 см3 азотной кислоты и кипятят до удаления окислов азота. Охлажденный раствор переносят в мерную колбу вместимостью 100 см3, доводят до метки водой и перемешивают.
Отбирают аликвотную часть раствора 5 см3 (при массовой доле никеля от 20 до 30 %), 7 см3 (при массовой доле никеля от 15 до 20 %) и 10,0 см3 (при массовой доле никеля свыше 10 % до 15 %), переносят в мерную колбу вместимостью 100 см3, приливают 10 см3 раствора калия-натрия виннокислого, 5 см3 раствора персульфата аммония, 5 см3 раствора едкого натра и 10 см3 диметилклиоксима. После прибавления каждого реактива раствор перемешивают, доводят до метки водой и снова перемешивают.
Оптическую плотность измеряют через 15 минут на фотоколориметре со светофильтром, имеющим область пропускания в интервале длин волн от 460 до 480 нм или на спектрофотометре при длине волны 460 нм, в кювете с толщиной слоя 5 мм относительно раствора сравнения.
Раствор сравнения готовят следующим образом: в мерную колбу вместимостью 100 см3 помещают 2 см3 стандартного раствора А и добавляют все реактивы в том же количестве, доводят водой до метки и перемешивают.
18.3.1. Построение градуировочного графика для массовой доли никеля от 10,1 до 30,0 %.
В мерные колбы вместимостью 100 см3 помещают стандартный раствор А в количестве 2 см3 и стандартный раствор Б в количестве 0,5; 2,0; 4,0; 6,0; 8,0; 10,0 см3, что соответствует 10,25; 11,0; 12,0; 13,0; 20,0; 30,0 % никеля при конечных навесках 0,01; 0,007 и 0,005 г соответственно. В каждую колбу приливают 10 см3 раствора калия-натрия виннокислого и далее анализ ведут, как указано в п. 18.3.
В раствор сравнения вводят 2 см3 стандартного раствора А и добавляют все реактивы в том же количестве.
18.4. Обработка результатов
18.4.1. Массовую долю никеля в процентах находят по градуировочному графику.
18.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 10).
Таблица 10
|
Абсолютные допускаемые расхождения, % |
|
|
От 10,1 до 15,0 |
0,15 |
|
Св. 15,0 " 20,0 |
0,25 |
|
" 20,0 " 25,0 |
0,30 |
|
" 25,0 " 30,0 |
0,35 |
19. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ ТИТАНА (0,005 - 3,5 %) С ДИАНТИПИРИЛМЕТАНОМ
19.1. Сущность метода
Метод основан на образовании окрашенного в желтооранжевый цвет комплексного соединения титана с диантипирилметаном в кислой среде с максимумом светопоглощения 390 нм. Оптимальный интервал кислотности от 0,5 до 0,8 н по соляной кислоте. Чувствительность метода 0,000001 г в 100 см3 раствора. Интенсивность окрашивания пропорциональна содержанию титана. Алюминий, марганец, медь, цирконий, кобальт, никель не образуют с диантипирилметаном окрашенных соединений и не мешают определению. Влияние железа, хрома и ванадия устраняют прибавлением аскорбиновой кислоты.
19.2. Аппаратура, реактивы и растворы
Фотоколориметр типа ФЭК-56 или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота серная по ГОСТ 4204-77, разбавленная 1:4.
Кислота азотная по ГОСТ 4461-77.
Кислота соляная по ГОСТ 3118-77, 1 н раствор.
Кислота аскорбиновая по ГОСТ 4815-76, 5 %-ный свежеприготовленный раствор.
Диантипирилметан, 5 %-ный раствор в 1 н соляной кислоте, свежеприготовленный.
19.3. Проведение анализа
Навеску стали массой 0,5 г (при массовой доле титана до 0,2 %), 0,25 г (при массовой доле титана свыше 0,2 до 0,4 %) и 0,1 г (при массовой доле титана свыше 0,4 %) помещают в коническую колбу вместимостью 250 см3, приливают 25 см3 серной кислоты, разбавленной 1:4 и растворяют при нагревании. По окончании растворения прибавляют по каплям азотную кислоту до прекращения вспенивания и выпаривают до начала выделения паров серной кислоты. Операцию выпаривания повторяют дважды. Сложные стали растворяют в царской водке, затем приливают 15 см3 серной кислоты, разбавленной 1:1 и выпаривают дважды до выделения паров серной кислоты. Содержимое стакана охлаждают, приливают 30 см3 воды и нагревают до растворения солей. Охлажденный раствор переносят в мерную колбу вместимостью 100 см3, доводят водой до метки и перемешивают. При анализе чугунов часть раствора отфильтровывают.
Аликвотную часть раствора 5 см3 помещают в мерную колбу вместимостью 100 см3, приливают 10 см3 раствора аскорбиновой кислоты и выдерживают мин. от 5 до 7. Затем прибавляют 10 см3 соляной кислоты плотностью 1,19 г/см3 и 5 см3 диантипирилметана. Раствор доливают до метки водой и перемешивают.
Через 50 мин измеряют оптическую плотность на фотоколориметре, со светофильтром, имеющим область пропускания в интервале длин волн от 400 до 450 нм или на спектрофотометре при длине волны 390 нм, в кювете с толщиной слоя 10 - 50 мм.
Раствором сравнения служит аликвотная часть раствора, к которой приливают все реактивы, за исключением раствора диантипирилметана.
19.4. Обработка результатов
19.4.1. Массовую долю титана (X) в процентах по методу сравнения вычисляют по формуле
![]()
где C - массовая доля титана в стандартном образце или в стандартном растворе, г;
Д1 - оптическая плотность анализируемого раствора;
Д2 - оптическая плотность стандартного раствора;
m - масса навески, г.
19.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных в (табл. 11).
Таблица 11
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,005 до 0,008 |
0,003 |
|
Св. 0,008 " 0,015 |
0,004 |
|
" 0,015 " 0,030 |
0,006 |
|
" 0,030 " 0,060 |
0,009 |
|
" 0,060 " 0,10 |
0,012 |
|
" 0,10 " 0,50 |
0,015 |
|
" 0,50 " 0,70 |
0,020 |
|
" 0,70 |
0,025 |
20. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ ТИТАНА
(0,1 - 3,0 %) В CTAЛЯX, НЕ
СОДЕРЖАЩИХ ВАНАДИЙ
20.1. Сущность метода
Метод основан на образовании окрашенного в желтый цвет соединения титана с перекисью водорода в сернокислой среде. Интенсивность окрашивания пропорциональна содержанию титана. Чувствительность метода составляет 0,000015 г в 50 мл раствора. Мешающее влияние железа устраняют прибавлением фосфорной кислоты, которая образует с ним бесцветное комплексное соединение, но в то же время несколько снижает интенсивность окраски титана. Поэтому прибавление излишнего количества фосфорной кислоты не рекомендуется.
Молибден с перекисью водорода в этих условиях так же дает окрашенное в желтый цвет соединение, которое по интенсивности слабее окраски надтитановой кислоты. Его влияние компенсируют введением такого же количества в стандартный образец. Влияние ионов обладающих собственной окраской (никеля, кобальта, меди, хрома), устраняют компенсацией с применением в качестве раствора сравнения части анализируемого образца без добавления перекиси водорода.
20.2. Аппаратура, реактивы и растворы
Фотоколориметр или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота серная по ГОСТ 4204-77, разбавленная 1:4.
Кислота азотная по ГОСТ 4461-77.
Кислота ортофосфорная по ГОСТ 6552-80, разбавленная 1:4.
Пергидроль по ГОСТ 10929-76, 3 %-ный раствор, свежеприготовленный.
20.3. Проведение анализа
Навеску массой 0,5 г помещают в коническую колбу вместимостью 250 см3, приливают 40 см3 серной кислоты, разбавленной 1:4 и растворяют при нагревании. По окончании растворения прибавляют по каплям азотную кислоту до прекращения вспенивания и выпаривают до выделения паров серной кислоты. Раствор охлаждают, разбавляют водой и нагревают для растворения выпавших солей. Охлажденный раствор переводят в мерную колбу вместимостью 100 см3, доводят водой до метки и перемешивают.
Аликвотную часть раствора 25 см3 помещают в мерную колбу вместимостью 50 см3, прибавляют 4 см3 фосфорной кислоты, разбавленной 1:4, 7 см3 перекиси водорода, доводят до метки водой и перемешивают.
Оптическую плотность измеряют на фотоколориметре со светофильтром, имеющим область пропускания в интервале длин волн от 400 до 450 нм или на спектрофотометре при длине волны 400 нм, в кювете с толщиной слоя 30 или 50 мм относительно раствора сравнения.
В качестве раствора сравнения применяют аликвотную часть раствора, к которой приливают все реактивы, за исключением раствора перекиси водорода.
20.4. Обработка результатов
20.4.1. Массовую долю титана (X) в процентах по методу сравнения вычисляют по формуле, приведенной в п. 19.4.1.
20.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 12).
Таблица 12
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,1 до 0,5 |
0,015 |
|
Св. 0,5 " 0,7 |
0,020 |
|
" 0,7 |
0,025 |
21. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ МЕДИ В УГЛЕРОДИСТЫХ И НИЗКОЛЕГИРОВАННЫХ СТАЛЯХ
Метод основан на образовании внутрикомплексного соединения меди с диэтилдитиокарбоматом натрия, окрашенного в желтокоричневый цвет. Развитие окраски происходит в аммиачной среде. Максимум светопоглощения находится при λ = 440 нм. Реакция образования комплекса очень чувствительна и может быть применена для определения следов меди. Интенсивность окрашивания пропорциональна массовой доле меди. Мешающее влияние железа, алюминия устраняют прибавлением лимонной кислоты или ее солей.
21.2. Аппаратура, реактивы и растворы
Фотоколориметр ФЭК-56 или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота серная по ГОСТ 4204-77, разбавленная 1:4.
Кислота азотная по ГОСТ 4461-77, разбавленная 1:1.
Аммиак водный по ГОСТ 3760-79.
Аммоний лимоннокислый однозамещенный по ГОСТ 7234-79, 25 %-ный раствор.
Желатина пищевая по ГОСТ 11293-78, 0,5 %-ный свежеприготовленный раствор.
Диэтилдитиокарбамат натрия по ГОСТ 8864-71, 0,1 %-ный свежеприготовленный раствор; готовят следующим образом: 0,1 г реактива помещают в коническую колбу вместимостью 200 см3, приливают 100 см3 аммиака водного, разбавленного 1:1, перемешивают и фильтруют через фильтр «белая лента».
Стандартный раствор меди; готовят следующим образом: 0,5 г металлической меди особой чистоты по ГОСТ 546-67 растворяют в 20 см3 азотной кислоты при нагревании. Раствор охлаждают, переводят в мерную колбу вместимостью 1 л, разбавляют водой до метки и перемешивают.
1 см3 раствора содержит 0,0005 г меди.
Навеску стали массой 0,5 г помещают в коническую колбу вместимостью 200 см3, приливают 20 см3 азотной кислоты, растворяют при нагревании и кипятят до удаления окислов азота. Раствор охлаждают, переводят в мерную колбу вместимостью 200 см3, доливают водой до метки и тщательно перемешивают.
Отбирают аликвотную часть раствора 10 см3 и переносят ее в мерную колбу вместимостью 100 см3, приливают 15 см3 раствора аммония лимоннокислого, 15 см3 раствора аммиака. К охлажденному раствору приливают 10 см3 раствора желатины, 10 см3 раствора диэтилдитикарбамата натрия, доливают водой до метки и перемешивают.
Оптическую плотность измеряют на фотоколориметре со светофильтром, имеющим область пропускания в интервале от 380 до 450 нм или на спектрофотометре при длине волны 440 нм, в кювете с толщиной слоя 20 мм.
Раствор для сравнения готовят следующим образом: в мерную колбу вместимостью 100 см3 помещают 10 см3 анализируемого раствора (вторую аликвотную часть), приливают 15 см3 раствора аммония лимоннокислого, 15 см3 раствора аммиака. К охлажденному раствору приливают 10 см3 раствора желатины, доливают водой до метки и перемешивают.
21.3.1. Построение градуировочного графика
В шесть колб вместимостью 200 см3 помещают шесть навесок железа массой 0,5 г. В пять колб приливают стандартный раствор меди в количестве 1,0; 2,0; 3,0; 4,0 и 5,0 см3, что соответствует 0,1; 0,2; 0,3; 0,4; 0,5 % меди. Во все колбы приливают по 20 см3 азотной кислоты и проводят анализ, как указано в п. 21.3.
Шестая навеска железа служит для приготовления раствора сравнения с добавлением всех реактивов.
По найденным значениям оптической плотности строят градуировочный график.
21.4. Обработка результатов
21.4.1. Массовую долю меди в процентах находят непосредственно по градуировочному графику.
21.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 13).
Таблица 13
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,01 до 0,02 |
0,005 |
|
Св. 0,02 " 0,05 |
0,008 |
|
" 0,05 " 0,10 |
0,01 |
|
" 0,10 " 0,25 |
0,02 |
|
" 0,25 " 0,50 |
0,03 |
22. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ МЕДИ В ЛЕГИРОВАННЫХ СТАЛЯХ
22.1. Сущность метода указана в п. 21.1
22.2. Аппаратура, реактивы и растворы указаны в п. 21.2.
22.3. Проведение анализа
Навеску стали массой 0,5 г помещают в коническую колбу вместимостью 200 см3, приливают 50 см3 серной кислоты, разбавленной 1:4 и растворяют при нагревании. После полного растворения прибавляют по каплям азотную кислоту по ГОСТ 4461-77 до прекращения вспенивания и кипятят до удаления окислов азота. Полученный раствор охлаждают, переводят в мерную колбу вместимостью 200 см3 и перемешивают.
Отбирают аликвотную часть раствора 10 см3 и переносят ее в мерную колбу вместимостью 100 см3, прибавляют 30 см3 раствора лимоннокислого аммония, 35 см3 раствора аммиака. К охлажденному раствору приливают 10 см3 раствора желатины, 10 см3 раствора диэтилдитиокарбомата натрия, доводят до метки водой и перемешивают.
Оптическую плотность измеряют на фотоколориметре со светофильтром, имеющим область пропускания в интервале от 380 до 450 нм или на спектрофотометре при длине волны 440 нм, в кювете с толщиной слоя 20 мм.
Раствор для сравнения готовят следующим образом: в мерную колбу вместимостью 100 см3 помещают 10 см3 анализируемого раствора (вторую аликвотную часть), приливают 30 см3 раствора лимоннокислого аммония, 35 см3 раствора аммиака. К охлажденному раствору приливают 10 см3 раствора желатины, доводят до метки водой и перемешивают.
22.4. Обработка результатов
22.4.1. Массовую долю меди (X) в процентах по методу сравнения вычисляют по формуле:
![]()
где C - массовая доля меди в стандартном образце, г;
Д1 - оптическая плотность анализируемого раствора;
Д2 - оптическая плотность стандартного раствора;
m - масса навески, г.
22.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 13).
23. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ ЦИРКОНИЯ
23.1. Сущность метода
Метод основан на образовании цветной реакции циркония с арсеназо Ш. Определение циркония проводят на фоне всех компонентов стали или чугуна в растворе 2 н соляной кислоты после восстановления трехвалентного железа гидроксиламином. Интенсивность окрашивания пропорциональна массовой доле циркония. Чувствительность метода 0,000005 г циркония в 50 см3 раствора.
При растворении навески стали существенное значение имеет выбор окислителя. Более надежным является пергидроль, которая легко разрушается при кипячении. Упаривание раствора досуха приводит к потере циркония в результате его гидролиза.
Для устранения влияния хрома, никеля, титана фотометрирование проводят против такого же раствора, содержащего трилон Б, маскирующий цирконий.
23.2. Аппаратура, реактивы и растворы
Спектрофотометр или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота соляная по ГОСТ 3118-77, разбавленная 1:1 и 2 н раствор.
Пергидроль по ГОСТ 10929-76.
Гидроксиламин солянокислый по ГОСТ 5456-80, 10-ный раствор.
Арсеназо Ш, 0,1 %-ный водный раствор.
Стандартный раствор циркония; готовят следующим образом: 0,3532 г хлорокиси циркония растворяют в 20 см3 соляной кислоты, переводят в мерную колбу вместимостью 1 л, доводят до метки водой и перемешивают. 1 см3 раствора содержит 0,0001 г циркония.
Навеску стали массой 0,25 г (при массовой доле циркония от 0,01 до 0,1 %) или 0,1 г (при массовой доле циркония от 0,1 до 0,5 %) помещают в стакан вместимостью 100 см3, приливают 30 см3 соляной кислоты, разбавленной 1:1 и растворяют при нагревании. После растворения прибавляют по каплям от 1 до 2 см3 пергидроля для окисления карбидов и кипятят до ее разрушения. Раствор охлаждают, переносят в мерную колбу вместимостью 100 см3, доводят до метки раствором 2 н соляной кислоты и перемешивают. При наличии в растворе графита, его отфильтровывают через фильтр средней плотности в сухую колбу или стакан, первые порции отбрасывают.
Отбирают аликвотную часть раствора 10 см3 и переносят ее в мерную колбу вместимостью 50 см3, приливают 10 см3 воды, 2 см3 раствора гидроксиламина и нагревают до кипения, для перевода циркония в однородную форму ионов и восстановления железа. Раствор охлаждают, добавляют 20 см3 раствора 2 н соляной кислоты, 1 см3 арсеназо Ш, доводят до метки той же кислотой и перемешивают.
Оптическую плотность измеряют на спектрофотометре при длине волны 650 нм или на фотоколориметре со светофильтром, имеющим область пропускания в интервале длин волн от 630 до 660 нм, в кювете с толщиной слоя 30 мм.
В качестве раствора сравнения используют вторую аликвотную часть анализируемого раствора, приготовленного в тех же условиях и содержащего 1 см3 раствора трилона Б для связывания циркония. Раствор трилона Б приливают перед добавлением арсеназо Ш.
23.4. Построение градуировочных графиков
23.4.1. Построение градуировочного графика для массовой доли циркония от 0,01 до 0,1 %.
В пять стаканов вместимостью 100 см3 помещают навеску железа или стали не содержащей циркония, равную анализируемой пробе, и 0,5; 1,0; 1,5; 2,0; 2,5 см3 стандартного раствора циркония, что соответствует 0,02; 0,04; 0,06; 0,08; 0,10 % циркония при навеске массой 0,25 г и аликвотной части раствора 10 см3. Содержимое стакана растворяют в 30 см3 соляной кислоты, разбавленной 1:1 и проводят анализ, как указано в п. 23.3. Шестой стакан служит для приготовления раствора сравнения, который проводят через все стадии анализа.
23.4.2. Построение градуировочного графика для определения массовой доли циркония от 0,1 до 0,5 %.
В пять стаканов вместимостью 100 см3 помещают навеску карбонильного железа или стали не содержащей циркония, равную анализируемой пробе, и 1,5; 2,0; 2,5; 3,0; 4,0; 5,0 см3 стандартного раствора циркония, что соответствует 0,15; 0,20; 0,25; 0,30; 0,40; 0,50 % циркония при навеске массой 0,1 г и аликвотной части раствора 10 см3. Содержимое стакана растворяют в 30 см3 соляной кислоты, разбавленной 1:1 и проводят анализ, как указано в п. 23.3. Шестой стакан служит для приготовления раствора сравнения, который проводят через все стадии анализа.
По полученным значениям оптической плотности строят градуировочный график.
23.5. Обработка результатов
23.5.1. Массовую долю циркония в процентах находят по градуировочному графику.
23.5.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 14).
Таблица 14
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,01 до 0,02 |
0,004 |
|
Св. 0,02 " 0,05 |
0,007 |
|
" 0,05 " 0,10 |
0,008 |
|
" 0,10 " 0,25 |
0,015 |
|
" 0,25 " 0,50 |
0,025 |
24. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ МОЛИБДЕНА (0,05 - 1,0 %) В СЛАБОЛЕГИРОВАННЫХ СТАЛЯХ
Метод основан на образовании окрашенного в оранжевожелтый цвет комплексного соединения пятивалентного молибдена с роданистыми солями в сернокислой среде. Восстановление шестивалентного молибдена производится тиомочевиной в присутствии ионов меди в качестве катализатора. Трехвалентное железо в присутствии тиомочевины восстанавливается до двухвалентного, которое не образует с роданидами окрашенных соединений.
Вольфрам до 0,005 г и ванадий до 0,002 г в конечном объеме не мешают определению молибдена. Наличие ванадия свыше 0,002 г компенсируют введением такого же количества в раствор сравнения.
Образующийся комплекс имеет максимум светопоглощения при длине волн от 440 до 470 нм. Чувствительность метода составляет 0,00001 г в 100 см3 раствора. Интенсивность окрашивания пропорциональна массовой доле молибдена.
24.2. Аппаратура, реактивы и растворы
Фотоколориметр или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота серная по ГОСТ 4204-77, разбавленная 1:4.
Кислота азотная по ГОСТ 4461-77.
Кислота соляная по ГОСТ 3118-77.
Смесь кислот; готовят следующим образом: 400 см3 серной кислоты по ГОСТ 4204-77 осторожно вливают в 1450 см3 воды и прибавляют 100 см3 соляной кислоты по ГОСТ 3118-77.
Медь сернокислая по ГОСТ 4165-78, 1 %-ный раствор.
Аммоний лимоннокислый по ГОСТ 9264-79, 30 %-ный раствор.
Тиомочевина по ГОСТ 6344-73, 5 %-ный свежеприготовленный раствор.
Калий роданистый по ГОСТ 4139-75, 50 %-ный раствор.
24.3. Проведение анализа
Навеску стали массой 0,2 г помещают в коническую колбу вместимостью 250 см3, приливают 40 см3 серной кислоты, разбавленной 1:4 и растворяют при нагревании. После полного растворения прибавляют по каплям азотную кислоту до прекращения вспенивания. Раствор выпаривают до появления паров серной кислоты. После охлаждения споласкивают стенки колбы водой и повторяют выпаривание. Выпаривание следуют вести осторожно во избежание выпадения нерастворимых основных солей. Раствор охлаждают. Выпавшие соли при охлаждении растворяют в 30 - 40 см3 воды при нагревании. Охлажденный раствор переводят в мерную колбу вместимостью 100 см3, доводят до метки водой и перемешивают.
Отбирают аликвотную часть раствора 10 см3 и переносят ее в мерную колбу вместимостью 100 см3, приливают 35 см3 смеси кислот, 1 см3 раствора сернокислой меди, 10 см3 раствора тиомочевины и выдерживают 5 минут. Затем приливают 2 см3 раствора роданистого калия, доводят до метки водой и перемешивают. Для полного восстановления железа и молибдена и развития окраски раствор выдерживают один час, после чего измеряют оптическую плотность на фотоколориметре, со светофильтром, имеющим область пропускания в интервале длин волн от 440 до 470 нм или на спектрофотометре при длине волны 440 нм, в кювете с толщиной слоя 50 мм.
В качестве раствора сравнения используют стандартный образец стали, близкой по химическому составу к анализируемому, но не содержащей молибдена.
24.4. Обработка результатов
24.4.1. Массовую долю молибдена (X) в процентах по методу сравнения вычисляют по формуле
![]()
где C - массовая доля молибдена в стандартном образце, г;
Д1 - оптическая плотность анализируемого раствора;
Д2 - оптическая плотность стандартного раствора;
m - масса навески, г.
24.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 15).
Таблица 15
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,05 до 0,10 |
0,012 |
|
Св. 0,10 " 0,50 |
0,020 |
|
" 0,50 " 1,00 |
0,035 |
|
" 1,00 |
0,05 |
25. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ
МОЛИБДЕНА
(0,5 - 3,0 %) В ВЫСОКОЛЕГИРОВАННЫХ СТАЛЯХ
25.1. Сущность метода указана в п. 24.1.
25.2. Аппаратура, реактивы и растворы указаны в п. 24.2.
25.3. Проведение анализа
Навеску стали массой 0,1 г помещают в коническую колбу вместимостью 200 см3, приливают 40 см3 серной кислоты, разбавленной 1:4 и растворяют при нагревании. При содержании в стали вольфрама для ускорения растворения прибавляют 10 см3 соляной кислоты плотностью 1,19 г/см3.
Если на дне колбы имеется значительное количество неразложившихся карбидов, раствор выпаривают до паров серной кислоты, охлаждают, осторожно приливают от 20 до 30 см3 воды, растворяют соли при нагревании и только после этого окисляют азотной кислотой, затем вторично выпаривают до паров серной кислоты.
Сплавы и стали сложного состава растворяют в смеси кислот (30 см3 соляной кислоты и 10 см3 азотной кислоты), после чего добавляют в раствор 20 см3 серной кислоты, разбавленной 1:1 и выпаривают дважды до паров серной кислоты. Выпаривание следует вести осторожно во избежании выпадения нерастворимых основных солей хрома.
Выпавшие во время выпаривания соли растворяют в 30 - 40 см3 воды (при наличии вольфрама приливают 10 см3 раствора лимоннокислого аммония) и осторожно раствор нагревают. Быстрое нагревание может привести к обугливанию лимоннокислого аммония.
Охлажденный раствор переводят в мерную колбу вместимостью 100 см3, доводят до метки водой и перемешивают.
Отбирают аликвотную часть раствора 5 см3 и переносят ее в мерную колбу вместимостью 100 см3, приливают 35 см3 смеси кислот, 1 см3 раствора сернокислой меди, 10 см3 раствора тиомочевины, 2 см3 раствора роданистого калия. Хорошо перемешивают после добавления каждого реактива и выдерживают 5 минут. После этого доводят до метки водой и перемешивают. Для полного восстановления железа и развития окраски выдерживают растворы от 35 до 40 минут.
Оптическую плотность измеряют на фотоколориметре со светофильтром, имеющим область пропускания в интервале длин волн от 440 до 470 нм или на спектрофотометре при длине волны 440 нм, в кювете с толщиной слоя 20 мм.
В качестве раствора сравнения используют вторую аликвотную часть анализируемого раствора, приготовленного в тех же условиях без добавления раствора роданистого калия.
25.4. Обработка результатов
25.4.1. Массовую долю молибдена (X) в процентах по методу сравнения вычисляют по формуле, указанной в п. 24.4.1.
25.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 15).
26. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ ВАНАДИЯ С ФЕНИЛАНТРАНИЛОВОЙ КИСЛОТОЙ
26.1. Сущность метода
Метод основан на образовании комплексного соединения при окислении фенилантраниловой кислоты раствором пятивалентного ванадия. Окраска устойчива в течение 30 мин и пропорциональна концентрации ванадия в пределах от 0,00001 до 0,0001 г в 50 см3 раствора. Комплекс образуется в среде более, чем 5М серной кислоты.
26.2. Аппаратура, реактивы и растворы
Фотоколориметр или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота серная по ГОСТ 4204-77, разбавленная 1:1.
Кислота ортофосфорная по ГОСТ 6552-80, разбавленная 1:4.
Смесь кислот; готовят следующим образом: к 40 см3 воды приливают 20 см3 серной кислоты плотностью 1,82 г/см3 и 20 см3 ортофосфорной кислоты плотностью 1,7 г/см3.
Кислота азотная по ГОСТ 4461-77.
Пергидроль по ГОСТ 10929-76.
Калий марганцовокислый по ГОСТ 20490-75, 0,3 %-ный раствор.
Натрий азотистокислый по ГОСТ 4168-79, 4 %-ный раствор.
Мочевина по ГОСТ 6691-77, 50 %-ный раствор.
Фенилантраниловая кислота, 0,1 %-ный раствор; готовят следующим образом: 0,1 г кислоты и 0,1 г безводного углекислого натрия растворяют при нагревании в 100 см3 воды. Готовят в день применения.
26.3. Проведение анализа
Навеску стали массой 0,2 г помещают в коническую колбу вместимостью 250 см3, приливают 25 см3 смеси кислот и растворяют при нагревании. После полного растворения прибавляют по каплям азотную кислоту до прекращения вспенивания. Раствор выпаривают до выделения густых паров серной кислоты, которые должны выделяться в течение от 5 до 7 минут. Затем раствор охлаждают, разбавляют водой, добавляют от 3 до 5 капель пергидроля, кипятят и снова охлаждают. Раствор переносят в мерную колбу вместимостью 200 см3, доводят до метки водой и перемешивают.
Отбирают аликвотную часть раствора от 5 до 10 см3 (в зависимости от массовой доли ванадия) и переносят ее в мерную колбу вместимостью 50 см3, прибавляют по каплям раствор марганцовокислого калия для окисления ванадия до розовой окраски, устойчивой в течение 5 минут. Затем вводят по каплям раствор азотистокислого натрия до исчезновения розового окрашивания, (тщательно перемешивают после добавления каждой капли) и вводят одну каплю в избытке. Сразу же приливают 1 см3 раствора мочевины для связывания избытка нитрита натрия, 5 см3 ортофосфорной кислоты и перемешивают, затем осторожно, не перемешивая, приливают 0,5 см3 раствора фенилантраниловой кислоты, быстро доводят до метки серной кислотой, разбавленной 1:1 и осторожно без сильного взбалтывания перемешивают, сразу охлаждают в струе холодной воды.
Оптическую плотность измеряют на фотоколориметре со светофильтром, имеющим область пропускания в интервале длин волн от 530 до 540 нм или на спектрофотометре при длине волны 536 нм, в кювете с толщиной слоя 20 мм.
В качестве раствора сравнения используют аликвотную часть анализируемого раствора без добавления фенилантраниловой кислоты.
Параллельно с анализом пробы проводят холостой опыт, в качестве которого используют массу навески карбонильного железа, проведенную через все стадии анализа.
При обработке результатов величину оптической плотности холостого опыта вычитают из величины оптической плотности анализируемого образца.
26.4. Обработка результатов
26.4.1. Массовую долю ванадия (X) в процентах по методу сравнения вычисляют по формуле
![]()
где C - массовая доля ванадия в стандартном образце, г;
Д1 - оптическая плотность анализируемого раствора;
Д2 - оптическая плотность стандартного раствора;
m - масса навески, г.
26.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 16).
Таблица 16
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,02 до 0,05 |
0,008 |
|
Св. 0,05 " 0,10 |
0,010 |
|
" 0,10 " 0,50 |
0,025 |
|
" 0,50 " 1,00 |
0,08 |
27. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ ВАНАДИЯ В ВИДЕ ФОСФОРНОВОЛЬФРАМОВАНАДИЕВОГО КОМПЛЕКСА
27.1. Сущность метода
Метод основан на образовании окрашенного в желтый цвет фосфорновольфрамованадиевого комплексного соединения (λ = 413 нм) в кислой среде. Мешающее влияние железа устраняют прибавлением фосфорной кислоты.
27.2. Аппаратура, реактивы и растворы
Фотоколориметр ФЭК-56 или другие приборы подобного типа обеспечивающие такую же точность измерения.
Кислота серная по ГОСТ 4204-77, разбавленная 1:9.
Кислота азотная по ГОСТ 4461-77.
Калий марганцовокислый по ГОСТ 20490-75, 4 %-ный раствор.
Натрий азотистокислый по ГОСТ 4168-79, 10 %-ный раствор.
Аммоний ванадиевокислый мета по ГОСТ 9336-75.
Железо.
Стандартные растворы ванадия А и Б.
Раствор А; готовят следующим образом: 0,4593 г ванадиевокислого аммония растворяют в 200 см3 воды, добавляют от 2 до 4 капель 25 %-ного раствора аммиака, переводят раствор в мерную колбу вместимостью 500 см3, разбавляют водой до метки и перемешивают.
1 см3 раствора А содержит 0,0004 г ванадия.
Раствор Б; готовят следующим образом: раствор А разбавляют водой в десять раз.
1 см3 раствора Б содержит 0,00004 г ванадия.
Навеску стали массой 0,2 г помещают в колбу вместимостью 100 см3 и растворяют в 15 см3 серной кислоты при умеренном нагревании. После полного растворения навески, приливают по каплям азотную кислоту до прекращения вспенивания раствора. Раствор выпаривают до появления паров серной кислоты. Содержимое стакана охлаждают, приливают 30 см3 воды и нагревают до растворения солей. К охлажденному раствору прибавляют раствор марганцовокислого калия до образования устойчивой розовой окраски. Через мин 3 - 5 избыток марганцовокислого калия восстанавливают раствором азотистокислого натрия, прибавляя его медленно no одной капле.
Затем приливают в колбу 5 см3 фосфорной кислоты, нагревают раствор до кипения, прибавляют 10 см3 раствора вольфрамовокислого натрия, выдерживают раствор от 80 до 90 °C в течение от 2 до 3 мин и фильтруют через фильтр «красная лента». Фильтрат собирают в мерную колбу вместимостью 100 см3. Фильтр промывают от 4 до 5 раз небольшим количеством горячей воды, собирая промывные воды в ту же колбу. Затем раствор охлаждают, доливают водой до метки и перемешивают.
Оптическую плотность окрашенного в желтый цвет раствора измеряют на фотоколориметре со светофильтром, имеющим область пропускания в интервале длин волн от 400 до 420 нм или на спектрофотометре при длине волны 413 нм, в кювете с толщиной слоя 30 мм. Раствором сравнения служит проба анализируемого материала, проведенная через все стадии анализа, которая содержит все реактивы, за исключением вольфрамовокислого натрия.
27.3.1. Построение градуировочного графика
Шесть навесок железа массой по 0,2 г помещают в колбы вместимостью по 100 см3. К пяти навескам приливают стандартный раствор Б в количестве 1,0; 2,0; 3,0; 4,0 и 5,0 см3, что соответствует, массовой доле ванадия 0,02; 0,04; 0,06; 0,08 и 0,10 %. В колбы приливают по 15 см3 серной кислоты и растворяют железо при умеренном нагревании. Далее анализ проводят, как указано в п. 27.3.
Шестая навеска используется для приготовления раствора сравнения с добавлением всех указанных реактивов.
По найденным значениям оптической плотности строят градуировочный график.
27.4. Обработка результатов
27.4.1. Массовую долю ванадия в процентах находят по градуировочному графику.
27.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 17).
Таблица 17
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,02 до 0,050 |
0,008 |
|
Св. 0,05 " 0,10 |
0,010 |
28. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ АЛЮМИНИЯ
28.1. Сущность метода
Метод основан на цветной реакции алюминия с эриохромцианином, окрашивающей раствор в ярко-красный цвет (λ = 535 нм) при pH = 6,0. Чувствительность метода составляет 0,0000005 г алюминия в 50 см3 раствора. Интенсивность окрашивания пропорциональна массовой доле алюминия. Мешающее влияние большинства элементов стали устраняют путем переведения алюминия в алюминат с одновременным осаждением гидроокисей мешающих элементов. Вольфрам не мешает в больших количествах. Влияние молибдена и ванадия при их массовой доле в стали свыше 1 % устраняют введением такого же количества в раствор сравнения.
28.2. Аппаратура, реактивы и растворы
Фотоколориметр ФЭК-56 и другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота соляная по ГОСТ 3118-77, разбавленная 1:1 и 1:25.
Кислота азотная по ГОСТ 4461-77.
Натр едкий по ГОСТ 4328-77, 20 %-ный раствор.
Аммиак водный по ГОСТ 3760-79, разбавленный 1:10.
Метиловый красный, 0,1 %-ный спиртовый раствор.
Эриохромцианин; готовят следующим образом: 0,375 г реактива растворяют в 100 см3 воды, прибавляют 12,5 г хлористого натрия, 12,5 г азотнокислого аммония, 1 см3 азотной кислоты плотностью 1,4 г/см3, переносят в мерную колбу вместимостью 500 см3, доводят водой до метки и перемешивают.
Ацетатный буферный раствор; готовят следующим образом: 320 г уксуснокислого аммония растворяют в воде, приливают 5 см3 98 %-ной уксусной кислоты, переводят в мерную колбу вместимостью 1 л, доводят до метки водой и перемешивают, pH от 6 до 6,5.
28.3. Проведение анализа
Навеску стали массой 0,5 г (при массовой доле алюминия от 0,005 до 0,2 %), 0,25 г (при массовой доле алюминия свыше 0,2 до 0,5 %) и 0,1 г (при массовой доле алюминия свыше 0,5 до 1 %) растворяют в стакане вместимостью от 250 до 300 см3 в 30 см3 соляной кислоты, разбавленной 1:1. После полного растворения прибавляют по каплям азотную кислоту плотностью 1,40 г/см3 до прекращения вспенивания и упаривают раствор до небольшого объема. Содержимое стакана переносят в полиэтиленовый сосуд, в который предварительно вносят 60 см3 едкого натра, тщательно перемешивают, доливают водой до 200 см3 и помещают в водяную баню, воду в которой нагревают до кипения и выдерживают в течение 30 мин, периодически перемешивая растворы.
Растворы с осадком гидроокисей охлаждают, переводят в мерную колбу вместимостью 250 см3, доводят до метки водой, перемешивают и переносят в полиэтиленовый сосуд, где проводилось осаждение. Часть раствора отфильтровывают в сухую колбу через фильтр «красная лента», первые порции фильтрата отбрасывают.
Аликвотную часть раствора 5 см3 переносят в стакан вместимостью 50 см3, приливают от 1 до 2 капель метилового красного и нейтрализуют соляной кислотой, разбавленной 1:1, затем раствором аммиака, разбавленным 1:10 и соляной кислотой, разбавленной 1:25 до изменения цвета от одной капли.
Содержимое стакана переносят в мерную колбу вместимостью 50 см3, приливают 5 см3 эриохромцианина, 5 см3 буферного раствора (отбирают только грушей), доводят до метки водой и перемешивают.
Оптическую плотность окрашенных растворов измеряют на фотоколориметре со светофильтром, имеющим область пропускания в интервале длин волн от 530 до 540 нм или на спектрофотометре при длине волны 535 нм, в кювете с толщиной слоя 10 мм.
В качестве раствора сравнения используют все необходимые реактивы, исключая аликвотную часть раствора.
Если в стали присутствуют ванадий и молибден свыше 1,0 %, то в раствор сравнения вводят такое же количество этих элементов.
Результаты анализа вычисляют методом сравнения со стандартным образом, близким по составу к анализируемой пробе и проведенным через все стадии анализа.
28.4. Обработка результатов
28.4.1. Массовую долю алюминия (X) в процентах по методу сравнения вычисляют по формуле
![]()
где C - массовая доля молибдена в стандартном образце или в стандартном растворе, г;
Д1 - оптическая плотность анализируемого раствора;
Д2 - оптическая плотность стандартного раствора;
m - масса навески, г.
28.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 18).
Таблица 18
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,005 до 0,02 |
0,001 |
|
Св. 0,02 " 0,10 |
0,002 |
|
" 0,10 " 0,50 |
0,005 |
|
" 0,50 " 1,00 |
0,025 |
|
" 1,00 |
0,050 |
29. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ ВОЛЬФРАМА (0,01 - 0,15 %)
29.1. Сущность метода
Метод основан на образовании в кислой среде в присутствии восстановителя, вольфрамовороданидного комплекса с максимумом светопоглощения от 390 до 450 нм.
Для определения малых количеств вольфрамовой кислоты проводят ее концентрирование на продуктах гидролиза - ниобиевой кислоты и перекиси марганца одновременно.
Осадок обрабатывают щелочью с целью перевода в раствор вольфрамовой кислоты, где и определяют роданидным методом.
29.2. Аппаратура, реактивы и растворы
Фотоколориметр ФЭК-56 или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота соляная плотностью 1,11 г/см3; готовят следующим образом: 200 см3 воды смешивают со 150 см3 соляной кислоты плотностью 1,19 г/см3.
Кислота азотная плотностью 1,2 г/см3, готовят следующим образом: 340 см3 воды смешивают с 240 см3 азотной кислоты плотностью 1,4 г/см3.
Кислота азотная по ГОСТ 4461-77.
Марганец сернокислый по ГОСТ 435-77, 7 %-ный раствор.
Калий марганцовокислый по ГОСТ 20490-75, 4 %-ный раствор.
Аммоний азотнокислый по ГОСТ 22867-77, 1 %-ный раствор.
Натр едкий по ГОСТ 4328-77, 10 %-ный раствор.
Калий роданистый по ГОСТ 4139-75, 25 %-ный раствор.
Олово двухлористое по ГОСТ 36-78, 10 %-ный раствор; готовят следующим образом: 25 г двухлористого олова растворяют в 250 см3 соляной кислоты плотностью 1,19 г/см3.
Титан треххлористый по ГОСТ 311-78 или приготовленный из двуокиси титана; 1,6 г двуокиси титана сплавляют с калием пиросернокислым при температуре от 800 до 850 °C. Плав выщелачивают в 250 см3 соляной кислоты, разбавленной 1:1 при нагревании без кипячения. Раствор отфильтровывают.
Перед началом определения часть раствора восстанавливают металлическим цинком до фиолетового окрашивания раствора.
Раствор ниобия с титром 0,001 г/см3, готовят следующим образом: 0,2 г металлического ниобия сплавляют с 5 г калия пиросернокислого, плав выщелачивают в 100 см3 10 %-ного раствора винной кислоты. Раствор переводят в мерную колбу вместимостью 200 см3, доводят до метки водой и перемешивают.
Стандартный раствор вольфрама готовят следующим образом: навеску ферровольфрама (стандартный образец № 202) массой 0,1414 г сплавляют с 5 г калия пиросернокислого при 800 °C, выщелачивают в 50 см3 10 %-ного раствора едкого натра, переводят в мерную колбу вместимостью 250 см3, доводят до метки водой и перемешивают. Щелочной раствор сразу фильтруют в полиэтиленовый сосуд, где и хранят. 1 см3 раствора содержит 0,0004 г вольфрама.
Навеску стали массой 1 г помещают в стакан вместимостью 300 см3 и растворяют при нагревании в смеси кислот: 10 см3 соляной кислоты плотностью 1,11 г/см3 и 20 см3 азотной кислоты плотностью 1,2 г/см3. По окончании растворения вводят 5 см3 раствора ниобия, 5 см3 раствора сернокислого марганца и выпаривают почти досуха. Приливают 10 см3 азотной кислоты плотностью 1,4 г/см3 и повторяют выпаривание почти досуха. Снова добавляют 10 см3 той же кислоты и упаривают до влажных солей. Все операции по выпариванию следует проводить очень осторожно. Выпавшие соли растворяют в 20 см3 азотной кислоты с уд. в. 1,2 г/см3, приливают 100 см3 кипящей воды, вводят калий марганцовокислый (4 см3 на каждый 0,1 г хрома и от 2 до 3 см3 в избытке).
Содержимое стакана кипятят, сохраняя общий объем 100 см3 раствора добавлением горячей воды до метки, предварительно нанесенной на стакан с помощью мерной колбы. После этого горячий раствор фильтруют через фильтр «белая лента» и промывают осадок на фильтре горячим 1 %-ным раствором азотнокислого аммония. Осадок вместе с фильтром переносят в стакан, где проводилось осаждение, прибавляют 20 см3 раствора едкого натра, немного воды и тщательно перемешивают стеклянной палочкой. Нагревают раствор с фильтром до кипения и кипятят в течение 2 минут.
Раствор охлаждают, доводят до метки водой, тщательно перемешивают палочкой, фильтруют через два фильтра «белая лента», первые порции фильтрата отбрасывают.
Отбирают аликвотную часть раствора 25 см3, помещают в мерную колбу вместимостью 50 см3, приливают 3 см3 раствора роданистого калия (аммония), 12 см3 раствора хлористого олова и охлаждают в проточной воде. Добавляют 2 см3 раствора треххлористого титана, доводят до метки водой и перемешивают.
Оптическую плотность измеряют на фотоколориметре со светофильтром, имеющим область пропускания в интервале длин волн от 390 до 450 нм или на спектрофотометре при длине волны 390 нм, в кювете с толщиной слоя 30 мм.
В качестве раствора сравнения применяют аликвотную часть раствора анализируемой пробы без раствора роданистого калия.
Массовую долю вольфрама находят по градуировочному графику.
29.3.1. Построение градуировочного графика
В пять стаканов вместимостью 300 см3 помещают навеску железа, равную анализируемой пробе и 0,5; 1,0; 2,0; 3,0; 4,0 см3 стандартного раствора вольфрама, что соответствует 0,02; 0,04; 0,08; 0,12; 0,16 % вольфрама при навеске массой 1 г и аликвотной части раствора пробы 25 см3. Содержимое стакана растворяют в смеси кислот и далее анализ ведут, как указано в п. 29.3.
29.4. Обработка результатов
29.4.1. Массовую долю вольфрама в процентах находят по калибровочному графику.
29.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 19).
Таблица 19
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,01 до 0,05 |
0,003 |
|
Св. 0,05 " 0,10 |
0,005 |
|
" 0,10 " 0,50 |
0,015 |
|
" 0,50 " 1,00 |
0,030 |
|
" 1,00 " 3,00 |
0,050 |
30. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ ВОЛЬФРАМА (СВЫШЕ 0,15 %)
30.1. Сущность метода
Метод основан на реакции образования роданидного комплекса пятивалентного вольфрама, окрашенного в желто-зеленый цвет, без предварительного отделения его от сопутствующих компонентов. При растворении и окислении металлического вольфрама и его карбидов в растворе, содержащем ортофосфорную кислоту, вольфрам образует комплексную растворимую фосфорновольфрамовую кислоту, которая не выпадает в осадок. В качестве восстановителя целесообразно употреблять раствор треххлористого титана, который более активен по сравнению с другими восстановителями. Восстановление происходит в сильнокислой среде, окраска развивается в течение от 5 до 12 мин. Восстановлению вольфрама должна предшествовать операция восстановления железа хлористым оловом. При такой последовательности восстановления железа и вольфрама устраняется возможность выпадания осадка фосфорнокислого титана.
Влияние ванадия свыше 0,2 % и хрома свыше 2 % можно устранить методом компенсации (введением в раствор сравнения соответствующих количеств этих элементов).
Наличие молибдена до 3 % не мешает определению вольфрама.
При наличии в стали мышьяка и сурьмы вводят двойное количество треххлористого титана.
30.2. Аппаратура, реактивы и растворы
Фотоколориметр ФЭК-56 или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Смесь кислот; готовят следующим образом: к 625 см3 воды приливают 250 см3 ортофосфорной кислоты плотностью 1,7 г/см3 и 150 см3 серной кислоты плотностью 1,82 г/см3.
Кислота азотная по ГОСТ 4461-77.
Олово двухлористое по ГОСТ 36-78, 20 %-ный раствор; готовят следующим образом: 50 г двухлористого олова растворяют в 250 см3 соляной кислоты плотностью 1,19 г/см3.
Титан треххлористый; готовят, как указано в п. 29.2.
Калий (аммоний) роданистый, 50 %-ный раствор.
30.3. Проведение анализа
Навеску стали массой 0,5 г (при массовой доле вольфрама до 1,0 %), 0,2 г (при массовой доле вольфрама свыше 1,0 % до 10,0 %) и 0,1 г (при массовой доле вольфрама свыше 10,0 %) помещают в коническую колбу вместимостью 250 см3 и растворяют в 40 см3 смеси кислот при нагревании. После полного растворения навески, приливают по каплям азотную кислоту до прекращения вспенивания. Раствор выпаривают до появления паров серной кислоты.
При анализе стали, содержащей большие количества хрома и никеля, навеску растворяют в смеси кислот, а затем добавляют 10 см3 соляной кислоты плотностью 1,19 г/см3 и 10 см3 азотной кислоты плотностью 1,4 г/см3, затем упаривают раствор до выделения густых паров серной кислоты. Дают раствору охладиться, обмывают стенки колбы водой и вновь охлаждают.
Охлажденный раствор переносят в мерную колбу вместимостью 250 см3, доводят водой до метки и перемешивают,
Аликвотную часть раствора 5 см3 помещают в мерную колбу вместимостью 50 см3, прибавляют 2 см3 раствора роданистого калия и несколько капель раствора хлористого олова (из предварительно отмеренных мензуркой 20 см3) до обесцвечивания, затем вводят 2 см3 раствора треххлористого титана, перемешивают раствор и после этого прибавляют основное количество раствора хлористого олова. Содержимое колбы перемешивают, доводят до метки водой и снова перемешивают.
Через 5 мин измеряют оптическую плотность на фотоколориметре, со светофильтром, имеющим область пропускания в интервале длин волн от 420 до 430 нм или на спектрофотометре при длине волны 420 нм, в кювете с толщиной слоя от 20 до 30 мм.
В качестве раствора сравнения используют аликвотную часть анализируемого раствора, в которую добавляют все реактивы, кроме раствора роданистого калия.
30.4. Обработка результатов
30.4.1. Массовую долю вольфрама (X) в процентах по методу, сравнения вычисляют по формуле
![]()
где C - массовая доля вольфрама в стандартном образце, г;
Д1 - оптическая плотность анализируемого раствора;
Д2 - оптическая плотность стандартного раствора;
m - масса навески, г.
30.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 19).
31. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ КОБАЛЬТА
31.1. Сущность метода
Метод определения кобальта основан на образовании с нитрозо-Р-солью растворимого в воде комплексного соединения, окрашивающего раствор в красный цвет. Комплекс образуется в среде с pH не ниже 6,5. Для создания среды применяют ацетатный буферный раствор. Окрашенный комплекс кобальта не образуется в сильнокислой среде. Это свойство использовано для приготовления раствора сравнения (сначала вводят кислоту, затем раствор нитрозо-Р-соли).
Ванадий, вольфрам и молибден при их массовой доле до 5 % значительного влияния не оказывают.
31.2. Аппаратура, реактивы и растворы
Фотоколориметр ФЭК-56 или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота соляная по ГОСТ 3118-77, разбавленная 1:1.
Кислота азотная по ГОСТ 4461-77, разбавленная 1:1.
Буферный раствор с pH 6,0 - 6,5; готовят следующим образом: 23,2 г уксуснокислого натрия по ГОСТ 199-78 растворяют в 1 л воды, добавляют 0,45 см3 98 %-ой уксусной кислоты, раствор перемешивают.
Нитрозо-Р-соль по ГОСТ 10553-75, 0,5 %-ный раствор.
Стандартный раствор кобальта; готовят следующим образом: 5,173 г соли азотнокислого кобальта растворяют в 100 см3 воды, переводят в мерную колбу вместимостью 1 л, доводят водой до метки и перемешивают.
1 см3 раствора содержит 0,001 г кобальта.
Навеску стали массой 0,2 г помещают в стакан вместимостью 100 см3 и растворяют в 20 см3 соляной кислоты, разбавленной 1:1. По окончании растворения приливают по каплям азотную кислоту до прекращения вспенивания, после чего раствор кипятят и выпаривают до сиропообразного состояния.
В стакан приливают 30 см3 воды и нагревают до растворения солей, после чего раствор охлаждают и переносят в мерную колбу вместимостью 200 см3, доводят до метки водой и перемешивают.
Аликвотную часть раствора 5 см3 помещают в мерную колбу вместимостью 100 см3, приливают 30 см3 ацетатного буферного раствора, 5 см3 нитрозо-Р-соли и кипятят раствор 1 мин, после этого добавляют 5 см3 азотной кислоты, разбавленной 1:1 и снова кипятят 1 мин. Раствор охлаждают, доводят до метки водой и перемешивают.
Оптическую плотность измеряют на фотоколориметре со светофильтром, имеющим область пропускания в интервале длин волн от 500 до 520 нм или на спектрофотометре при длине волны 520 нм, в кювете с толщиной слоя 10 мм.
Раствор для сравнения готовят следующим образом: в мерную колбу вместимостью 100 см3 помещают аликвотную часть анализируемого раствора 5 см3, приливают 30 см3 ацетатного буферного раствора, 5 см3 азотной кислоты, разбавленной 1:1, 5 см3 раствора нитрозо-Р-соли и нагревают до кипения.
После охлаждения доводят раствор до метки водой и перемешивают.
31.3.1. Построение калибровочного графика.
В шесть стаканов вместимостью 100 см3 помещают навеску железа или стали, не содержащей кобальта, равную анализируемой пробе, и 1,0; 2,0; 4,0; 6,0; 8,0; 10,0 см3 стандартного раствора кобальта, что соответствует 0,5; 1,0; 2,0; 3,0; 4,0; 5,0 % кобальта при навеске массой 0,2 г и аликвотной части раствора пробы 5 см3. Содержимое стакана растворяют в 30 см3 соляной кислоты, разбавленной 1:1, по окончании растворения прибавляют по каплям азотную кислоту до прекращения вспенивания, после чего раствор кипятят и выпаривают до сиропообразного состояния. Далее анализ ведут, как указано в п. 31.3.
По полученным значениям оптической плотности растворов строят градуировочный график.
31.4. Обработка результатов
31.4.1. Массовую долю кобальта в процентах находят по градуировочному графику.
31.4.2. Массовую долю кобальта (X) в процентах по методу сравнения вычисляют по формуле
![]()
где C - массовая доля кобальта в стандартном образце или в стандартном растворе, г;
Д1 - оптическая плотность анализируемого раствора;
Д2 - оптическая плотность стандартного раствора;
m - масса навески, г.
31.4.3. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 20).
Таблица 20
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,5 до 1,0 |
0,04 |
|
Св. 1,0 " 2,0 |
0,08 |
|
" 2,0 " 3,0 |
0,15 |
|
" 3,0 " 5,0 |
0,20 |
32. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ МЫШЬЯКА
32.1. Сущность метода
Метод определения мышьяка основан на восстановлении пятивалентного мышьяка гипофосфитом кальция, в присутствии катализатора хлорной меди, до элементарного. При добавлении раствора иодистого калия, элементарный мышьяк переходит в коллоидное состояние, окрашивающее раствор в желтый цвет. Трехвалентное железо предварительно восстанавливают хлористым оловом. При определении мышьяка навеску металла растворяют в азотной кислоте, разбавленной 1:1, или в смеси соляной и азотной кислот. Мышьяк после разложения пробы находится в растворе в нелетучей пятивалентной форме. При растворении в соляной и серной кислотах соединения мышьяка разлагаются, и мышьяк улетучивается в виде мышьяковистого водорода.
32.2. Аппаратура, реактивы и растворы
Фотоколориметр ФЭК-56 или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота азотная по ГОСТ 4461-77, разбавленная 1:1.
Кислота соляная по ГОСТ 3118-77, разбавленная 1:1.
Медь хлорная по ГОСТ 4167-74, 5 %-ный раствор в соляной кислоте, разбавленной 1:1.
Калий иодистый по ГОСТ 4232-74, насыщенный раствор в соляной; кислоте, разбавленной 1:1.
Олово двухлористое по ГОСТ 36-78, 20 %-ный раствор в соляной кислоте плотностью 1,19 г/см3.
Гипофосфит кальция по ГОСТ 11678-75, насыщенный раствор в соляной кислоте, разбавленной 1:1.
Растворы двухлористого олова и гипофосфита кальция используют свежеприготовленными.
Стандартный раствор мышьяка; готовят следующим образом: 0,132 г трехокиси мышьяка растворяют в 5 см3 едкого натра, 5 %-ного раствора, прибавляют 30 см3 воды, подкисляют соляной кислотой до слабокислой реакции, переводят в мерную колбу вместимостью 1 л, доводят до метки водой и перемешивают.
1 см3 раствора содержит 0,0001 г мышьяка.
Навеску стали массой 0,5 г (при массовой доле мышьяка от 0,01 до 0,1 %) или 0,1 г (при массовой доле мышьяка от 0,1 до 0,2 %) растворяют в 20 см3 смеси кислот: азотной - плотностью 1,40 г/см3 и соляной - плотностью 1,19 г/см3 в соотношении 1:2. По окончании растворения раствор выпаривают досуха для полного удаления азотной кислоты, сухой остаток растворяют в 10 см3 соляной кислоты, разбавленной 1:1, слегка подогревая (кипячение недопустимо). Если на дне стакана имеются частицы графита раствор фильтруют прямо в мерную колбу вместимостью 100 см3, фильтр промывают от 4 до 5 раз горячей соляной кислотой, разбавленной 1:1. К раствору в мерную колбу добавляют 1 см3 раствора хлорной меди и 10 см3 (при массе навески 0,5 г) или 5 см3 (при массе навески 0,1 г) раствора хлористого олова. Раствор перемешивают, затем вводят 10 см3 раствора гипофосфита кальция и снова перемешивают. После этого вводят 10 см3 раствора иодистого калия, осторожно перемешивают, доводят до метки соляной кислотой, разбавленной 1:1, нагревают до температуры от 50 до 60 °C и выдерживают в теплом месте в течение 35 - 40 мин. Содержимое колбы охлаждают и измеряют оптическую плотность растворов на фотоколориметре со светофильтром, имеющим область пропускания в интервале длин волн от 530 до 540 нм или на спектрофотометре при длине волны 540 нм, в кювете с толщиной слоя 20 мм.
В качестве раствора сравнения используют воду.
Массовую долю мышьяка определяют по градуировочному графику или методом сравнения со стандартным образцом, близким по составу к анализируемой пробе и проведенным через все стадии анализа.
32.3.1. Построение градуировочного графика
В шесть стаканов вместимостью 100 см3 помещают навески массой от 0,1 до 0,5 г железа или низкоуглеродистой стали, не содержащей мышьяка в соответствии с массой и навески анализируемой пробы. В пять стаканов приливают последовательно 0,5; 1,0; 1,5; 2,0 и 2,5 см3 стандартного раствора мышьяковистокислого натрия, что соответствует 0,01; 0,02; 0,03; 0,04; 0,05 % мышьяка при навеске образца массой 0,5 г. Шестой стакан служит для приготовления контрольной пробы для учета массовой доли мышьяка в реактивах. Затем во все шесть стаканов приливают по 20 см3 смеси кислот азотной и соляной 1:2 и далее анализ проводят, как указано в п. 32.3.
По найденным значениям оптической плотности строят градуировочный график.
32.4. Обработка результатов
32.4.1. Массовую долю мышьяка в процентах находят по градуировочному графику.
32.4.2. Массовую долю мышьяка (X) в процентах по методу сравнения вычисляют по формуле
![]()
где C - массовая доля мышьяка в стандартном образце или в стандартном растворе, г;
Д1 - оптическая плотность анализируемого раствора;
Д2 - оптическая плотность стандартного раствора;
m - масса навески, г.
32.4.3. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 21).
Таблица 21
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,01 до 0,02 |
0,005 |
|
Св. 0,02 " 0,04 |
0,007 |
|
" 0,04 " 0,10 |
0,012 |
|
" 0,10 " 0,20 |
0,020 |
33. ЭКСТРАКЦИОННО-ФОТОМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ ТАНТАЛА
33.1. Сущность метода
Метод основан на образовании окрашенного соединения фторотанталата с бриллиантовым зеленым, экстрагируемого бензолом. Окраска стабилизируется добавлением ацетона и устойчива длительное время. Интенсивность окрашивания пропорциональна массовой доле тантала. Чувствительность метода составляет 0,000005 г в 25 см3 раствора. Определению мешают титан, цирконий, алюминий при их классовой доле свыше 5 %. Максимум светопоглощения находится при λ = 656 нм.
33.2. Аппаратура, реактивы и растворы
Фотоколориметр или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота азотная по ГОСТ 4461-77.
Кислота серная по ГОСТ 4204-77.
Кислота фтористоводородная (плавиковая) по ГОСТ 10484-78, 40 %-ный раствор.
Раствор для разбавления; готовят следующим образом: 20 г винной кислоты растворяют в 150 см3 воды, прибавляют 29,6 см3 серной кислоты плотностью 1,82 г/см3, переносят в мерную колбу вместимостью 1 л, доводят водой до метки и перемешивают.
Натрий фтористый по ГОСТ 4463-76, 5 %-ный раствор (хранят в полиэтиленовой посуде).
Бензол по ГОСТ 5955-75.
Ацетон по ГОСТ 3513-74.
Бриллиантовый зеленый, 0,5 %-ный раствор.
Стандартный раствор тантала А; готовят следующим образом: 0,1012 г тантала марки ТВЧ1 помещают в платиновую чашку и растворяют в смеси кислот (3 см3 фтористоводородной кислоты, 40 %-ного раствора, 2 см3 азотной кислоты плотностью 1,4 г/см3 и 5 см3 серной кислоты плотностью 1,82 г/см3), раствор упаривают до начала выделения паров серной кислоты, после чего чашку охлаждают, приливают 20 см3 5 %-ного раствора винной кислоты, переводят в мерную колбу вместимостью 1 л, доводят до метки тем же раствором винной кислоты и перемешивают.
1 см3 раствора содержит 0,0001 г тантала. Раствор хранят в полиэтиленовой посуде.
Раствор Б; готовят следующим образом: 10 см3 стандартного раствора А помещают в мерную колбу вместимостью 100 см3, доводят до метки водой и перемешивают.
1 см3 раствора Б содержит 0,00001 г тантала.
Навеску стали массой 0,5 г (при массовой доле тантала от 0,01 до 0,1 %) и 0,2 г (при массовой доле тантала свыше 0,1 %) помещают в платиновую чашку, приливают 5 см3 воды, 3 см3 плавиковой кислоты, 3 см3 серной кислоты плотностью 1,82 г/см3 и растворяют при нагревании. По окончании растворения окисляют азотной кислотой до прекращения вспенивания и кипятят до начала выделений паров серной кислоты. После охлаждения приливают 50 см3 раствора для разбавления, переносят содержимое чашки в мерную колбу вместимостью 100 см3, доводят до метки водой, перемешивают и переносят раствор в полиэтиленовый сосуд.
Отбирают аликвотную, часть раствора 5 см3, помещают в полиэтиленовый стакан, добавляют до 50 см3 раствора для разбавления, приливают 30 см3 бензола, 2 см3 бриллиантового зеленого, 10 см3 раствора фтористого натрия и перемешивают 1 минуту. Содержимое стакана переносят в делительную воронку и отделяют бензольный слой от водного. 15 см3 бензольного экстракта помещают в мерную колбу вместимостью 25 см3, разбавляют до метки ацетоном и измеряют оптическую плотность на фотоколориметре со светофильтром, имеющим область пропускания в интервале длин волн от 650 до 660 нм или на спектрофотометре при длине волны 656 нм, в кювете с толщиной слоя 20 мм.
Массовую долю тантала находят по градуировочному графику.
33.3.1. Построение градуировочного графика
Навеску стали, не содержащей тантала, массой 0,5 г помещают в платиновую чашку, приливают 5 см3 воды, 3 см3 плавиковой кислоты, 5 см3 серной кислоты, плотностью 1,82 г/см3 и растворяют при нагревании. После растворения прибавляют по каплям азотную кислоту до прекращения вспенивания и кипятят до начала выделения паров серной кислоты.
После охлаждения приливают 50 см3 раствора для разбавления, содержимое чашки переносят в мерную колбу вместимостью 100 см3, доводят до метки водой, перемешивают и переносят раствор в полиэтиленовый сосуд.
Аликвотную часть 5 см3 помещают в шесть полиэтиленовых стаканов, вводят последовательно 0,5; 1,0; 2,0; 3,0; 4,0 см3 стандартного раствора тантала с Т = 0,00001 г/см3 что соответствует 0,02; 0,04; 0,08; 0,12; 0,16 % тантала, приливают до 50 см3 раствора для разбавления, 30 см3 бензола и далее анализ ведут, как указано в п. 33.3.
Шестой стакан служит для приготовления раствора сравнения, который проводят через все стадии анализа.
По полученным значениям оптической плотности растворов строят градуировочный график.
33.4. Обработка результатов
33.4.1. Массовую долю тантала находят по градуировочному графику.
33.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 22).
Таблица 22
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,01 до 0,05 |
0,005 |
|
Св. 0,05 " 0,10 |
0,007 |
|
" 0,10 " 0,50 |
0,01 |
34. ЭКСТРАКЦИОННО-ФОТОМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ БОРА В УГЛЕРОДИСТЫХ И НИЗКОЛЕГИРОВАННЫХ СТАЛЯХ
34.1. Сущность метода
Метод основан на образовании комплексного соединения аниона ![]() с
бриллиантовым зеленым, экстрагируемого бензолом.
с
бриллиантовым зеленым, экстрагируемого бензолом.
Полное
переведение бора в ![]() достигается в кислой среде не ниже 0,7
н по серной кислоте.
достигается в кислой среде не ниже 0,7
н по серной кислоте.
Экстрагирование комплекса проводят при pH = 3 - 5.
Мешающее влияние элементов, образующих устойчивые фторидные комплексы, устраняют введением избытка фтористого аммония.
34.2. Аппаратура, реактивы и растворы
Фотоколориметр или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота серная по ГОСТ 4204-77, х.ч., о.с.ч., разбавленная 1:4.
Пергидроль по ГОСТ 10929-76.
Аммоний фтористый, 2М раствор; готовят следующим образом: 74 г соли растворяют в 200 - 300 см3 вода, переводят в мерную колбу вместимостью 1 л, доводят до метки водой и перемешивают. Хранят в полиэтиленовой посуде.
Уротропин, 40 %-ный раствор.
Бриллиантовый зеленый, 0,1 %-ный раствор; готовят следующим образом: 0,1 г реагента помещают в стакан вместимостью 100 см3, приливают от 60 до 70 см3 воды и периодически перемешивают стеклянной палочкой. Затем фильтруют в мерную колбочку, вместимостью 100 см3, доводят до метки водой и перемешивают. Раствором можно пользоваться на другой день при хранении в холодильнике и в течение от 4 до 5 суток.
Бензол по ГОСТ 5955-75.
Навеску стали массой 0,2 г помещают в стакан вместимостью 100 см3, приливают пипеткой 10 см3 серной кислоты, разбавленной 1:4 и, накрыв часовым стеклом, растворяют при нагревании. После полного растворения навески вводят пипеткой 1 см3 пергидроля для окисления карбидов, смывают стенки стакана водой и кипятят до полного разложения пергидроля, не допуская выделения паров серной кислоты. Раствор охлаждают и переносят в мерную колбу вместимостью 25 см3, доводят до метки водой и перемешивают.
Отбирают
аликвотную часть раствора 5 см3, переносят в платиновую чашку,
вводят 2 см3 раствора фтористого аммония, перемешивают и выдерживают
35 глин для образования аниона ![]() . Устанавливают необходимое для
нейтрализации количество уротропина. С этой целью отбирают пипеткой 5 см3
раствора холостого опыта, помещают в
стеклянный стаканчик емкостью 50 см3, добавляют от 1 до 2 капель
бриллиантового зеленого и нейтрализуют уротропином, из пипетки с делениями, до
перехода окраски из желтой в голубовато-зеленую.
. Устанавливают необходимое для
нейтрализации количество уротропина. С этой целью отбирают пипеткой 5 см3
раствора холостого опыта, помещают в
стеклянный стаканчик емкостью 50 см3, добавляют от 1 до 2 капель
бриллиантового зеленого и нейтрализуют уротропином, из пипетки с делениями, до
перехода окраски из желтой в голубовато-зеленую.
Установленное
количество уротропина приливают ко всем пробам, и по истечении 35 минут,
необходимых для полного образования аниона ![]() , раствор из платиновой чашки переносят в
делительную воронку, чашку споласкивают строго 10 см3 дистиллированной воды, приливают 5 см3
раствора бриллиантового зеленого, 10 см3 бензола (пипеткой с помощью
груши) и экстрагируют в течение одной минуты. После разделения фаз сливают
водный слой, сливая вместе с ним от 0,5 до 1,0 см3 бензольного
экстракта. Органический слой помещают в сухой стакан вместимостью 50 см3,
затем в кювету с толщиной слоя 5 мм и сразу замеряют оптическую плотность на
фотоколориметре со светофильтром, имеющим область пропускания в интервале от
600 до 610 нм или на спектрофотометре при длине волны 610 нм.
, раствор из платиновой чашки переносят в
делительную воронку, чашку споласкивают строго 10 см3 дистиллированной воды, приливают 5 см3
раствора бриллиантового зеленого, 10 см3 бензола (пипеткой с помощью
груши) и экстрагируют в течение одной минуты. После разделения фаз сливают
водный слой, сливая вместе с ним от 0,5 до 1,0 см3 бензольного
экстракта. Органический слой помещают в сухой стакан вместимостью 50 см3,
затем в кювету с толщиной слоя 5 мм и сразу замеряют оптическую плотность на
фотоколориметре со светофильтром, имеющим область пропускания в интервале от
600 до 610 нм или на спектрофотометре при длине волны 610 нм.
В качестве раствора сравнения используют бензол.
Параллельно с анализом пробы проводят холостой опыт, в качестве которого используют навеску стандартного образца, не содержащую бор и проведенную через все стадии анализа.
При обработке результатов величину оптической плотности холостого опыта вычитают из величины оптической плотности анализируемого образца.
34.4. Обработка результатов
34.4.1. Массовую долю бора (X) в процентах по методу сравнения вычисляют по формуле
![]()
где C - массовая доля бора в стандартном образце, г;
Д1 - оптическая плотность анализируемого раствора;
Д2 - оптическая плотность стандартного раствора;
m - масса навески, г.
34.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 23).
Таблица 23
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,001 до 0,003 |
0,0005 |
|
Св. 0,003 " 0,005 |
0,0006 |
|
" 0,005 " 0,008 |
0,0008 |
|
" 0,008 " 0,010 |
0,0012 |
35. ЭКСТРАКЦИОННО-ФОТОМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ БОРА В ВЫСОКОЛЕГИРОВАННЫХ СТАЛЯХ
35.1. Сущность метода
Метод
основан на образовании комплексного соединения аниона ![]() c бриллиантовым
зеленым, экстрагируемого бензолом. Мешающее влияние образуемых в данных
условиях фторидных комплексов других элементов, в частности железа, алюминия,
титана и циркония устраняют введением избытка раствора фтористого аммония.
Медь, молибден и ниобий не мешают определению.
c бриллиантовым
зеленым, экстрагируемого бензолом. Мешающее влияние образуемых в данных
условиях фторидных комплексов других элементов, в частности железа, алюминия,
титана и циркония устраняют введением избытка раствора фтористого аммония.
Медь, молибден и ниобий не мешают определению.
Определение бора в сложнолегированных сталях затруднено из-за плохой растворимости основы металла, а также карбидных и боридных фаз. Для полного переведения бора в раствор навеску стали растворяют при слабом нагревании с обратным воздушным холодильником.
35.2. Аппаратура, реактивы и растворы указаны в п. 34.2.
Навеску стали массой 0,5 г помещают в коническую или круглую с плоским дном колбу вместимостью 200 см3 (предварительно проверенную на содержание в ней бора), приливают пипеткой 20 см3 серной кислоты, разбавленной 1:4 и ведут растворение с воздушным холодильником при слабом нагревании. После полного разложения навески добавляют гашеткой 2 см3 пергидроля для окисления карбидов, ополаскивают стенки колбы водой и кипятят до разложения пергидроля. Промывают холодильник водой, охлаждают раствор и переводят в мерную колбу вместимостью 50 см3, доводят до метки водой и перемешивают.
Аликвотную часть раствора 5 см3 переносят в платиновую чашку и далее проводят анализ, как описано в п. 34.3.
35.4. Массовую долю бора (X) в процентах по методу сравнения вычисляют по формуле, приведенной в п. 34.4.1.
35.4.1. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 23).
36. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ НИОБИЯ (0,005 - 0,1 %) В СТАЛЯХ, НЕ СОДЕРЖАЩИХ МОЛИБДЕН И ВАНАДИЙ
Метод основан на цветной реакции ниобия с сульфохлорфенолом С, окрашивающей раствор в синефиолетовый цвет (λ = 656 нм), в среде 1,5 н соляной кислоты в присутствии винной кислоты, препятствующей гидролизу ниобия. Для улучшения стабильности оптической плотности вводят ацетон (15 % по объему). Интенсивность окрашивания пропорциональна массовой доле ниобия. Чувствительность метода составляет 0,000002 г в 50 см3 раствора. Мешающее влияние трехвалентного железа устраняют, восстанавливая его до двухвалентного с помощью гидроксиламина. При наличии в образце циркония, в раствор вводят трилон Б; влияние вольфрама устраняют, связывая его в растворимый комплекс винной кислотой.
36.2. Аппаратура, реактивы и растворы
Фотоколориметр ФЭК-56 или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота винная по ГОСТ 5817-77.
Кислота соляная по ГОСТ 3118-77, разбавленная 1:1.
Кислота азотная по ГОСТ 4461-77.
Гидроксиламин солянокислый по ГОСТ 5456-80, 20 %-ный раствор.
Ацетон по ГОСТ 2603-71.
Трилон Б по ГОСТ 10659-73, 2 %-ный раствор.
Сульфохлорфенол С, ч.д.а., 0,1 %-ный раствор; готовят в день применения или пропускают через смолу КУ-2, который используют в течение двух недель.
Стандартный раствор ниобия; готовят следующим образом: 0,1 г металлического ниобия сплавляют с 5 г калия пиросернокислого при 700 °C в течение от 3 до 5 минут.
Плав выщелачивают в 8 %-ном растворе винной кислоты, охлаждают и переводят в мерную колбу вместимостью 1 л. До метки доводят тем же раствором винной кислоты и перемешивают.
1 мл раствора содержит 0,0001 г ниобия.
Навеску стали массой 0,5 г помещают в стакан вместимостью 250 см3, добавляют 3 г винной кислоты, 40 см3 соляной кислоты и растворяют при нагревании. После полного растворения прибавляют по каплям азотную кислоту до прекращения вспенивания и упаривают раствор до полного удаления окислов азота, но не допуская обугливания винной кислоты. Полученный раствор охлаждают, переводят в мерную колбу, вместимостью 100 см3 и перемешивают.
Отбирают аликвотную часть раствора 10 см3 и переносят в мерную колбу вместимостью 50 см3, приливают 10 см3 гидроксиламина выдерживают при температуре от 70 до 80 °C в течение от 5 до 10 мин для восстановления железа. К охлажденному раствору приливают 7,5 см3 ацетона, 12 см3 соляной кислоты, разбавленной 1:1, вводят 2 см3 сульфохлорфенола С, доливают до метки водой и перемешивают. Полученные растворы помещают в водяную баню, нагревают при температуре от 50 до 60 °C в течение от 5 до 10 мин, охлаждают и измеряют оптическую плотность на спектрофотометре при длине волны 656 нм или на фотоколориметре, со светофильтром, имеющим область пропускания в интервале длин волн от 650 до 660 нм, в кювете с толщиной слоя 20 мм.
В качестве раствора сравнения применяют стандартный образец, близкий по химическому составу к анализируемому, но не содержащий ниобий и проведенный через все стадии.
36.3.1. Построение градуировочного графика
В шесть стаканов вместимостью 250 см3 помещают навеску стандартного образца, не содержащего ниобий, равную анализируемой пробе и 0,25; 0,5; 1,0; 2,0; 5,0 см3 стандартного раствора ниобия, что соответствует 0,005; 0,01; 0,02; 0,04; 0,10 % ниобия при навеске массой 0,5 г и аликвотной части раствора пробы 10 см3, вносят 3 г винной кислоты и растворяют в 40 см3 соляной кислоты, разбавленной 1:1. После полного растворения прибавляют по каплям азотную кислоту до прекращения вспенивания, кипятят до удаления окислов азота и далее анализ ведут, как указано в п. 35.3.
Шестой стакан служит для приготовления раствора сравнения, который проводят через все стадии анализа.
По полученным значениям оптической плотности растворов строят градуировочный график.
36.4. Обработка результатов
36.4.1. Массовую долю ниобия находят по градуировочному графику.
36.4.2. Абсолютные допускаемые расхождения между результатами трех параллельных определений не должны превышать значений, указанных (в табл. 24).
Таблица 24
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,005 до 0,01 |
0,0008 |
|
Св. 0,01 " 0,05 |
0,004 |
|
" 0,05 " 0,10 |
0,006 |
|
" 0,10 " 0,5 |
0,15 |
|
" 0,5 " 1,0 |
0,025 |
|
" 1,0 |
0,05 |
37. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ НИОБИЯ
(свыше 0,1 %) В СТАЛЯХ, НЕ СОДЕРЖАЩИХ ВАНАДИЙ И МОЛИБДЕН
37.1. Сущность метода указана в п. 36.1.
37.2. Аппаратура, реактивы и растворы указаны в п. 36.2.
37.3. Проведение анализа
Навеску стаж массой 0,1 г помещают в стакан вместимостью 100 см3, добавляют 3 г винной кислоты, 20 см3 соляной кислоты, разбавленной 1:1 и растворяют при нагревании. После полного растворения прибавляют по каплям азотную кислоту до прекращения вспенивания и кипятят до полного разрушения карбидов и удаления окислов азота, но не допуская обугливания винной кислоты.
Если массовая доля кремния в образце превышает 1,5 %, осадок после растворения отфильтровывают и промывают несколько раз горячей водой. Фильтр с осадком помещают в фарфоровый тигель, высушивают, озоляют и прокаливают при температуре от 800 до 900 °C. Остаток в тигле сплавляют с 5 г калия пиросернокислого при температуре от 600 до 700 °C в течение от 3 до 5 мин. Плав выщелачивают в основном фильтрате от всей массы навески образца.
Полученный раствор охлаждают, переносят в мерную колбу вместимостью 250 см3, доводят до метки водой и перемешивают.
Отбирают аликвотную часть раствора 10 см3 (при массовой доле ниобия от 0,1 до 2 %) и 5,0 см3 (при массовой доле ниобия свыше 2 %) помещают в мерную колбу вместимостью 50 см3, приливают 5 см3 гидроксиламина и нагревают до температуры от 70 до 80 °C в течение от 3 до 5 мин для восстановления железа. К охлажденному раствору прибавляют 7,5 см3 ацетона и далее анализ ведут, как указано в п. 36.3,
В качестве раствора сравнения применяют стандартный образец, близкий по химическому составу к анализироемому, но не содержащий ниобий.
37.4. Обработка результатов
37.4.1. Массовую долю ниобия (X) в процентах по методу сравнения вычисляют по формуле
![]()
где C - массовая доля ниобия в стандартном образце, г;
Д1 - оптическая плотность анализируемого раствора;
Д2 - оптическая плотность стандартного раствора;
m - масса навески, г.
37.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 24).
38. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ НИОБИЯ
(0,005 - 0,1 %) В НИЗКОЛЕГИРОВАННЫХ СТАЛЯХ,
СОДЕРЖАЩИХ МОЛИБДЕН, ВАНАДИЙ И ВОЛЬФРАМ
38.1. Сущность метода указана в п. 36.1.
Метод основан на предварительном отделении ниобия от мешающих элементов путем осаждения его фениларсоновой кислотой. В качестве соосадителя применяют раствор циркония, влияние которого на окрашенный комплекс ниобия с сульфохлорфенолом С устраняют раствором трилона Б.
38.2. Аппаратура, реактивы и растворы
Фотоколориметр ФЭК-56 или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота соляная по ГОСТ 3118-77 и разбавленная 1:1.
Хлорокись циркония х.ч. или ч.д.а., 0,7 %-ный раствор; готовят следующим образом: 0,7 г соли растворяют в 10 см3 соляной кислоты, плотностью 1,19 г/см3, доводят объем водой до 100 см3 и перемешивают.
Кислота винная по ГОСТ 5817-77, 8 %-ный раствор.
Кислота азотная по ГОСТ 4461-77.
Кислота фениларсоновая, ч.д.а., ч, 5 %-ный раствор.
Калий пиросернокислый по ГОСТ 7172-76.
Трилон Б (натриевая соль этилендиаминтетрауксусной кислоты), 2 %-ный раствор.
Ацетон по ГОСТ 3513-74.
Сулъфохлорфенол С, 0,1 %-ный раствор; готовят в день применения или пропускают через смолу КУ-2, который используют в течение двух недель.
Стандартный раствор ниобия; готовят, как указано в п. 36.2.
Навеску стали массой 0,5 г помещают в стакан вместимостью 300 см3, вводят 5 см3 раствора хлорокиси циркония, приливают 40 см3 соляной кислоты и растворяют при нагревании. После полного растворения прибавляют по каплям азотную кислоту до прекращения вспенивания и кипятят до полного разрушения карбидов и удаления окислов азота. В полученный раствор приливают 25 см3 соляной кислоты, разбавленной 1:1, 40 см3 винной кислоты, разбавляют горячей водой до объема 200 см3 и нагревают до 90 °C, после чего вводят 25 см3 фениларсоновой кислоты и оставляют на ночь. Осадок отделяют на фильтре «синяя лента» и промывают горячим раствором азотнокислого аммония, подкисленным несколькими каплями азотной кислоты.
Промытый осадок вместе с фильтром помещают в фарфоровый тигель, озоляют и прокаливают от 800 до 900 °C в течение 45 минут. Остаток сплавляют с 3 г калия пиросернокислого от 700 до 750 °C. Тигли переносят в те же стаканы, в которых вели осаждение и выщелачивают в 70 см3 раствора винной кислоты. Охлажденный раствор переводят в мерную колбу вместимостью 100 см3, доводят до метки водой и перемешивают. Если раствор мутный, фильтруют.
Отбирают аликвотную часть раствора 10 см3 и переносят ее в мерную колбу вместимостью 50 см3, прибавляют 2,5 см3 трилона Б, 7,5 см3 ацетона, 12 см3 соляной кислоты, разбавленной 1:1, 2 см3 сульфохлорфенола С, доливают до метки водой и перемешивают. Полученные растворы помещают в водяную баню, нагревают при температуре от 50 до 60 °C в течение от 5 до 10 минут, охлаждают и измеряют оптическую плотность на спектрофотометре при длине волны 656 нм или на фотоколориметре, имеющим область пропускания в интервале длин волн от 650 до 660 нм, в кювете с толщиной слоя 20 мм.
В качестве раствора сравнения применяют стандартный образец, близкий по химическому составу к анализируемому, но не содержащий ниобия, проведенный через все стадии анализа с введением в него такого же количества раствора хлорокиси циркония.
38.3.1. Построение градуировочного графика
В шесть стаканов вместимостью 300 см3 помещают массу навески стандартного образца, не содержащего ниобий (с.о. 363), равную анализируемой пробе, и 0,25; 0,5; 1,0; 2,0; 5,0 см3 стандартного раствора ниобия, что соответствует 0,005; 0,01; 0,02; 0,04; 0,1 % ниобия при навеске массой 0,5 г и аликвотной части раствора 10 см3 вводят 5 см3 раствора хлорокиси циркония в каждый стакан, 40 см3 соляной кислоты, разбавленной 1:1 и далее анализ ведут, как указано в п. 38.3.
Шестой стакан служит для приготовления раствора сравнения, который проводят через все стадии анализа.
По полученным значениям оптической плотности строят градуировочный график.
38.4. Обработка результатов
38.4.1. Массовую долю ниобия находят по градуировочному графику.
38.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 24).
39. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ НИОБИЯ (СВЫШЕ 0,3 %) В СТАЛЯХ, СОДЕРЖАЩИХ МОЛИБДЕН, ВАНАДИЙ
39.1. Сущность метода указана в п. 36.1.
Метод основан на предварительном осаждении ниобия от мешающих элементов путем осаждения его фениларсоновой кислотой без соосадителя.
39.2. Аппаратура, реактивы и растворы
Фотоколориметр ФЭК-56 или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота соляная по ГОСТ 3118-77, разбавленная 1:1.
Кислота винная по ГОСТ 5817-77, 8 %-ный раствор.
Кислота азотная по ГОСТ 4461-77.
Кислота фениларсоновая, ч.д.а., ч, 5 %-ный раствор.
Калий пиросернокислый по ГОСТ 7172-76.
Ацетон по ГОСТ 3513-74.
Сульфохлорфенол С, ч.д.а., 0,1 %-ный раствор; готовят в день применения или пропускают через смолу КУ-2 и применяют в течение двух недель.
Навеску стали массой 0,5 г помещают в стакан вместимостью 300 см3, приливают 40 см3 соляной кислоты, разбавленной 1:1 и растворяют при нагревании. По окончании растворения прибавляют по каплям азотную кислоту до прекращения вспенивания и кипятят до полного разрушения карбидов и удаления окислов азота. В полученный раствор приливают 25 см3 соляной кислоты плотностью 1,19 г/см3, разбавляют горячей водой до объема 200 см3 и нагревают до 90 °C, после чего вводят 25 см3 раствора фениларсоновой кислоты и оставляют на ночь. Осадок отделяют на фильтре «синяя лента» и далее анализ ведут, как указано в п. 38.3.
Отбирают аликвотную часть раствора 10 см3 (при массовой доле ниобия от 0,1 до 1,0 %) и 5,0 см3 (при массовой доле ниобия свыше 1 %) помещают в мерную колбу вместимостью 50 см3, приливают 7,5 см3 ацетона, 12 см3 соляной кислоты, разбавленной 1:1 и далее анализ ведут, как указано в п. 38.3.
В качестве раствора сравнения применяют стандартный образец, близкий по химическому составу к анализируемому, но не содержащий ниобия и проведенный через все стадии анализа.
39.4. Обработка результатов
39.4.1. Массовую долю ниобия (X) в процентах по методу сравнения вычисляют по формуле, приведенной в п. 37.4.1.
39.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 24).
40. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ ЦЕРИЯ И ДРУГИХ РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ В УГЛЕРОДИСТЫХ И НИЗКОЛЕГИРОВАННЫХ СТАЛЯХ
Метод основан на цветной реакции редкоземельных элементов с арсеназо Ш, окрашивающей раствор в красно-фиолетовый цвет (λ = 660 нм) в слабокислой среде. Цветная реакция развивается сразу после добавления реагента и устойчива в течение суток. Интенсивность окрашивания пропорциональна массовой доле редкоземельных элементов. Чувствительность метода 0,0000025 г в 50 см3 раствора. Избирательность цветной реакции не высока - мешают железо, медь, ниобий, титан, хром, ванадий и др. элементы.
Мешающее влияние элементов устраняют осаждением редкоземельных элементов в виде оксалатов при pH 3, применяя в качестве коллектора хлористый кальций.
40.2. Аппаратура, реактивы и растворы
Спектрофотометр или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота соляная по ГОСТ 3118-77, разбавленная 1:1 и 0,01 н раствор.
Кислота азотная по ГОСТ 4461-77.
Кислота щавелевая по ГОСТ 22180-76, насыщенный раствор.
Аммиак водный по ГОСТ 3760-79.
Пергидроль по ГОСТ 10929-76.
Кальций хлористый по ГОСТ 4141-66; готовят следующим образом: 44 г хлористого кальция CaCl2 · 6Н2О растворяют в воде, переносят в мерную колбу вместимостью 1 л доводят водой до метки и перемешивают.
Арсеназо Ш по ТУ 6-09-4151-75, 0,04 %-ный раствор; готовят следующим образом: 0,2 г арсеназо Ш растворяют в 0,01 н соляной кислоте, переводят в мерную колбу емкостью 500 см3 и доводят до метки 0,01 н соляной кислотой.
Кислота аскорбиновая по ГОСТ 4815-76, 5 %-ный раствор.
Буферный раствор с pH 1,8; готовят следующим образом: 83 см3 0,2 н раствора соляной кислоты смешивают с 250 см3 0,2 н раствора, хлористого калия, переводят в мерную колбу вместимостью 1 л доводят до метки водой и перемешивают.
Стандартный раствор церия (лантана) А; готовят следующим образом: 1,443 г соли сернокислого церия 4-х водного помещают в термостойкий стакан вместимостью 250 см3, смачивают водой и приливают при перемешивании соляную кислоту по ГОСТ 3118-77 до полного растворения соли, выпаривают почти досуха, до полного удаления хлора, образовавшегося в результате окисления соляной кислоты сернокислым церием. Сухой остаток растворяют в 0,1 н соляной кислоте, переводят в мерную колбу вместимостью 500 см3, доводят до метки 0,1 н соляной кислотой и перемешивают.
1 см3 раствора содержит 0,001 г церия.
Раствор Б; готовят следующим образом: 5 см3 раствора А переносят в мерную колбу вместимостью 100 см3, доводят до метки раствором 0,01 н соляной кислоты и перемешивают.
1 см3 раствора содержит 0,00005 г церия.
Титр стандартного раствора устанавливают следующим образом: отбирают 3 аликвотные части раствора по 50 см3, помещают в стакан вместимостью 300 см3, приливают 50 см3 горячего раствора 1 %-ной щавелевой кислоты, по каплям аммиак водный, доводят pH до 3, подогревают до 80 °C и оставляют на ночь. Осадок отфильтровывают на фильтр «синяя лента», промывают от 5 до 6 раз 1 %-ной щавелевой кислотой, переносят в фарфоровый тигель, озоляют и прокаливают при ³ 800 °C до постоянного веса. Взвешивают в виде СеО2.
Титр стандартного раствора (Т), выраженный в г/см3 церия, вычисляют по формуле
![]()
где m - масса осадка в анализируемом образце, г;
0,8141 - коэффициент пересчета массы осадка окисла церия на церий;
V - объем раствора, взятый для анализа, см3.
Навеску стали массой 0,5 г (при массовой доле церия от 0,005 до 0,15 %) и 0,25 г (при массовой доле церия свыше 0,15 %) помещают в стакан вместимостью 300 см3, приливают 30 см3 соляной кислоты, разбавленной 1:1 и растворяют при нагревании. По окончании растворения прибавляют по каплям азотную кислоту до прекращения вспенивания и кипятят до небольшого объема. Разбавляют раствор водой до 150 см3, приливают 80 см3 щавелевой кислоты, 5 см3 раствора хлористого кальция и приливают по каплям аммиак водный до появления белой мути и в избытке от 1 до 2 капель. Полное осаждение оксалатов достигается при pH = 3 - 4, которую проверяют по индикаторной универсальной бумаге. Раствор с выпавшим осадком оставляют на ночь.
Осадок отделяют на фильтре «синяя лента», промывают раствором 1 %-ной щавелевой кислоты. Промытый осадок вместе с фильтром помещают в фарфоровый тигель, озоляют и прокаливают при температуре от 700 до 800 °C в течение от 30 до 45 мин. Тигли с прокаленным осадком охлаждают, осадок осторожно смачивают водой, приливают 5 см3 соляной кислоты плотностью 1,19 г/см3 и несколько капель пергидроля. Содержимое тигля количественно переносят в стакан вместимостью 100 см3 и упаривают досуха. Сухой остаток растворяют в 0,01 н растворе соляной кислоты, переносят в мерную колбу вместимостью 100 см3, доводят до метки той же кислотой и перемешивают.
Отбирают аликвотную часть раствора 10 см3 (при массовой доле церия от 0,005 до 0,07 %) и 5 см3 (при массовой доле церия свыше 0,07 %), помещают в мерную колбу вместимостью 50 см3, приливают 5 см3 раствора аскорбиновой кислоты, 5 см3 буферного раствора, 5 см3 арсеназо Ш, доводят до метки 0,01 н раствором соляной кислоты и перемешивают. Оптическую плотность измеряют на фотоколориметре со светофильтром, имеющим область пропускания в интервале длин волн от 650 до 660 нм или на спектрофотометре при длине волны 660 нм, в кювете с толщиной слоя 30 мм.
В качестве раствора сравнения применяют навеску стали, близкой по химическому составу к анализируемой, но не содержащей редкоземельных элементов и проведенной через все стадии анализа.
Результаты анализа вычисляют по градуировочному графику.
40.3.1. Построение градуировочного графика
В шесть стаканов вместимостью 300 см3 помещают массу навески железа или стали, не содержащей редкоземельных элементов, равную анализируемой пробе, и 0,5; 1,0; 2,0; 3,0; 4,0; 5,0 см3 стандартного раствора Б, что соответствует 0,005; 0,01; 0,02; 0,04; 0,06; 0,08 и 0,1 % церия при навеске массой 0,5 г и аликвотной части раствора пробы 10 см3. Содержимое стакана растворяют в 30 см3 соляной кислоты и далее анализ ведут, как указано в п. 39.3.
40.4. Обработка результатов
40.4.1. Массовую долю редкоземельных элементов от 0,005 до 0,1 % находят по графику, построенному для массовой доли от 0,005 до 0,1 % при навеске 0,5 г и аликвотной части раствора 10 см3.
Массовую долю редкоземельных элементов от 0,1 до 0,3 % (навеска массой 0,25 г, аликвотная часть раствора 5 см3) находят по этому же графику. Результат определения умножают на 4.
40.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 25).
Таблица 25
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,005 до 0,01 |
0,002 |
|
Св. 0,01 " 0,05 |
0,005 |
|
" 0,05 " 0,10 |
0,007 |
|
" 0,10 |
0,015 |
41. ФОТОКОЛОРИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ ЦЕРИЯ И ДРУГИХ РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ В ВЫСОКОЛЕГИРОВАННЫХ СТАЛЯХ
41.1. Сущность метода указана в п. 40.1.
Данный метод предусматривает осаждение аммиаком редкоземельных элементов, железа, титана, марганца и др. в виде гидроокисей в сернокислой среде, окислив предварительно хром и марганец до высших валентностей. Хром, никель, кобальт, частично вольфрам и ванадий при этом остаются в растворе.
41.2. Аппаратура, реактивы и растворы
Фотоколориметр или другие приборы подобного типа, обеспечивающие такую же точность измерения.
Кислота соляная по ГОСТ 3118-77, разбавленная 1:1 и 0,01 н раствор.
Кислота азотная по ГОСТ 4461-77.
Кислота серная по ГОСТ 4204-77.
Серебро азотнокислое по ГОСТ 1277-75, 5 %-ный раствор.
Аммоний надсернокислый (персульфат), 20 %-ный раствор, свежеприготовленный.
Аммиак водный по ГОСТ 3760-79.
Пергидроль по ГОСТ 10929-76.
Кислота щавелевая по ГОСТ 5.1173-71, насыщенный раствор.
Кальций хлористый по ТУ 6-09-4578-78 приготовление указано в п. 40.2.
Кислота аскорбиновая по ГОСТ 4815-76, 5 %-ный раствор.
Буферный раствор с pH 1,8; приготовление указано в п. 40.2.
Арсеназо Ш по ТУ 6-09-4151-75, 0,04 %-ный раствор; приготовление указано в п. 40.2.
Стандартный раствор церия (лантана); приготовление и установка титра указаны в п. 40.2.
41.3. Проведение анализа
Навеску стали массой 0,5 г (при массовой доле церия от 0,005 до 0,07 %) и 0,25 г (при массовой доле церия свыше 0,07 %) помещают в стакан вместимостью 300 см3 и растворяют при нагревании в 40 см3 соляной кислоты, разбавленной 1:1 или царской водке. По окончании растворения прибавляют по каплям азотную кислоту до прекращения вспенивания и кипятят до небольшого объема, после чего охлаждают, приливают 10 см3 серной кислоты плотностью 1,82 г/см3 и упаривают до выделения паров серной кислоты. Раствор охлаждают, осторожно разбавляют водой до 100 - 150 см3, прибавляют 5 см3 раствора азотнокислого серебра, 20 см3 раствора надсернокислого аммония и нагревают до полного окисления марганца. Длительное кипячение недопустимо. В теплые растворы добавляют по каплям аммиак водный до выпадения гидратов и небольшой избыток до сильного запаха. Раствор с выпавшим осадком выдерживают в теплом месте для коагуляции осадка, после чего фильтруют через фильтр «красная лента». Стакан обмывают над фильтром от 2 до 3 раз горячей водой, содержащей несколько капель аммиака водного и фильтр с осадком промывают от 5 до 6 раз этим же раствором. Осадок на фильтре растворяют в те же стаканы, где проводилось осаждение, горячим раствором соляной кислоты в присутствии пергидроля (в 1 л воды содержится 50 см3 соляной кислоты плотностью 1,19 г/см3 и 10 см3 пергидроля). После растворения осадка фильтр от 2 до 3 раз промывают горячей водой. Полученный раствор разбавляют до 150 см3 водой, приливают 80 см3 щавелевой кислоты, 5 см3 раствора хлористого кальция и добавляют по каплям аммиак водный до появления белой мути и в избытке от 1 до 2 капель. Полное осаждение оксалатов достигается при pH = 3 - 4 и контролируют по универсальной индикаторной бумаге. Далее анализ ведут, как указано в п. 40.3.
Результаты анализа вычисляют по градуировочному графику.
41.4. Обработка результатов
41.4.1. Массовую долю редкоземельных элементов находят по графику, построенному в условиях проведения анализа.
41.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 25).
42. ОПРЕДЕЛЕНИЕ АЗОТА МЕТОДОМ ОТГОНКИ В ВИДЕ АММИАКА
42.1. Сущность метода
Азот в железных сплавах содержится в твердом растворе и в связанном состоянии в виде химических соединений - нитридов ванадия, железа, алюминия и других элементов.
При растворении образца в соляной или серной кислотах азот твердого раствора и связанный (нитридный) азот вступает в реакцию с выделяющимся атомарным водородом и образует аммиак, а затем аммиачную соль соответствующей кислоты. При нагревании с концентрированными растворами щелочи в дистилляционной колбе аммонийная соль разлагается с выделением свободного аммиака, который отгоняют паром или сжатым воздухом в сосуд - приемник. После поглощения аммиака водой раствор оттитровывают 0,01 н серной кислотой в присутствии смешанного индикатора.
42.2. Аппаратура, реактивы и растворы
Установка для отгонки азота изготавливается по ТУ 25-11-489-70
Кислота серная по ГОСТ 4204-77, разбавленная 1:4.
Кислота ортофосфорная по ГОСТ 6552-80.
Кислота соляная по ГОСТ 3118-77.
Кислота винная по ГОСТ 5817-77.
Натр едкий по ГОСТ 4328-77, 30 %-ный раствор, прокипяченный.
Индикатор смешанный, готовят следующим образом: 0,062 г метилового красного и 0,041 г метиленового голубого растворяют в 100 см3 спирта.
Титрованный раствор серной кислоты 0,01 н раствор; готовят следующим образом: 2,86 см3 серной кислоты плотностью 1,82 г/см3 переносят в мерную колбу вместимостью 1 л, доливают до метки водой и перемешивают. Получают 0,1 н раствор. Для титрования его разбавляют в 10 раз.
0,1 н раствор серной кислоты можно приготовить из фиксанала, который также разбавляют в 10 раз. Получают 0,01 н титрованный раствор серной кислоты.
Титр раствора устанавливают по стандартному образцу стали или чугуна.
Для установки титра 0,01 н раствора серной кислоты берут стандартный образец, аналогичный по массовой доле азота и проводят через все стадии анализа.
Титр 0,01 н раствора серной кислоты (Т), выраженный в граммах азота, вычисляют по формуле
![]()
где C - массовая доля азота в стандартном образце, %;
m - масса навески стандартного образца, г;
V - объем раствора серной кислоты, израсходованный на титрование азота, содержащегося во взятой навеске стандартного образца с учетом холостого опыта, см3.
42.3. Проведение анализа
Навеску массой 3,0 г (при массовой доле азота от 0,005 до 0,025 %), 1,0 г (при массовой доле азота от 0,025 до 0,1 %) и 0,5 г (при массовой доле азота свыше 0,1 %) помещают в коническую колбу вместимостью 250 см3, приливают 40 см3 серной кислоты, разбавленной 1:4 и растворяют при нагреваний. По окончании растворения окисляют пергидролем.
Труднорастворимые сплавы типа ИЖС растворяют в 40 см3 серной кислоты, разбавленной 1:4 и 10 см3 фосфорной кислоты плотностью 1,70 г/см3. Доводят дважды до выделения паров серной кислоты. Содержимое колбы охлаждают. Соли растворяют в 20 - 30 см3 воды, приливают 20 см3 соляной кислоты, плотностью 1,19 г/см3 и продолжают нагревание. Так повторяют от 2 до 3 раз до исчезновения темных частиц на дне колбы, после этого производят окисление пергидролем.
Если в стали массовая доля кремния превышает 2 %, то в раствор перед окислением вводят от 1,5 до 2,0 г фтористого натрия до полного растворения кремневой кислоты.
После растворения, раствор переносят в колбу для перегонки, в которую предварительно приливают 100 см3 раствора едкого натра ополаскивают колбу, в которой велось растворение, и воронку водой (из предварительно отмеренных мензуркой 100 см3) и закрывают кран. Включают парообразователь и производят отгонку аммиака с паром, улавливая перегоняющийся аммиак в колбе-приемнике, в которую предварительно наливают 120 см3 воды и несколько капель индикатора (воду в приемнике нейтрализуют до кислой реакции от одной капли 0,01 н серной кислоты). Отгонку продолжают до получения объема от 240 до 250 см3.
Полученный раствор оттитровывают 0,01 н раствором серной кислоты до изменения окраски индикатора от зеленой до красно-фиолетовой.
Одновременно проводят холостой опыт на определение массовой доли азота в реактивах и воде, исключая пергидроль.
При обработке результатов объем серной кислоты, израсходованный на титрование холостого опыта вычитают из объема серной кислоты израсходованного на титрование анализируемого образца.
42.4. Обработка результатов
42.4.1. Массовую долю азота в процентах (1) вычисляют по формуле
![]()
где Т - титр раствора серной кислоты, выраженный в граммах азота;
V - объем раствора серной кислоты, израсходованный на титрование с учетом холостого опыта, см3;
m - масса навески, пробы, г.
Примечание:
Комната, где проводится определение азота, должна быть изолирована от помещения, где работают с азотной кислотой, аммиаком и аммонийными солями.
Посуда, применяемая при определении азота, должна быть тщательно вымыта и в дальнейшем употребляется по своему назначению.
Прибор для отгонки предварительно пропаривают в течение нескольких часов или проводят холостой отгон с несколькими миллилитрами щелочи.
42.4.2. Абсолютные допускаемые расхождения между результатами параллельных определений не должны превышать значений, указанных (в табл. 26).
Таблица 26
|
Абсолютные допускаемые расхождения, % |
|
|
От 0,005 до 0,01 |
0,001 |
|
Св. 0,01 " 0,02 |
0,0015 |
|
" 0,02 " 0,05 |
0,002 |
|
" 0,05 " 0,10 |
0,0035 |
|
" 0,10 " 0,20 |
0,010 |
43. ТРЕБОВАНИЯ БЕЗОПАСНОСТИ
43.1. Общие положения
43.1.1. Общая организация работы по технике безопасности в лаборатории возлагается на заведующего лабораторией, а по отдельным участкам на их руководителей. Заведующий лабораторией организует обучение и инструктирование работников лаборатории по безопасным методам работы.
43.1.2. Заведующий лабораторией не имеет право допускать к работе лиц, не получивших инструктаж и не ознакомленных с условиями работы. Повторный инструктаж проводится ежеквартально.
43.1.3. К работе в химической лаборатории допускаются лица не моложе 18 лет, прошедшие предварительный медосмотр и не имеющие противопоказаний, Повторный медосмотр проводится ежегодно.
43.1.4. Химические лаборатории должны быть расположены в помещениях с естественным освещением, отоплением, водопроводом и канализацией.
43.1.5. Во всех помещениях химической лаборатории должна быть приточно-вытяжная вентиляция, обеспечивающая нормальную очистку воздуха.
43.1.6. Все реактивы должны храниться в таре с надписью, категорически запрещается хранение каких-либо реактивов без наименований.
43.1.7. Все работы в химической лаборатории, должны проводиться при исправном состоянии электрооборудования, электропроводки и заземляющих устройств.
43.1.8. В каждом лабораторном помещении должны находиться огнетушитель, песок, кошма или одеяло.
43.1.9. В помещениях лаборатории запрещается:
загромождать коридоры, проходы и подходы к средствам пожаротушения;
убирать случайно пролитые огнеопасные жидкости при зажженных горелках и включенных электронагревательных приборах;
хранить и принимать пищу и молоко на рабочем месте;
43.1.10. При работе в вечернее и ночное время, а также при выполнении особо опасных работ в лаборатории должно находиться не менее двух человек, при этом один из них назначается старшим.
43.1.11. По окончании работы необходимо убрать все реактивы в надлежащее место, вымыть руки, закрыть газовые и водопроводные краны, отключить все нагревательные приборы, отключить общий рубильник, свет, лабораторию закрыть на ключ и сдать под охрану.
43.2. Требования безопасных условий труда
43.2.1. Каждый работающий в химической лаборатории должен знать:
свойства имеющихся в лаборатории химических реактивов, технических продуктов, продуктов реакции, а также веществ, поступающих в лабораторию на анализ, особенно их токсичность, огнеопасность и взрывоопасность;
опасные моменты при проведении работ и способы их предупреждения;
меры первой помощи при отравлениях, ожогах, поражениях электрическим током и т.д.
43.2.2. К работам, связанным с обслуживанием аппаратов под давлением, баллонов и сосудов со сжатыми и сжиженными газами, компрессоров допускаются лица после обучения и проверки знаний в квалификационной комиссии. Недопустимо, чтобы аппаратура и детали, присоединяемые к кислородному баллону, имели хотя бы следы масла. Это может привести к взрыву.
43.2.3. Все работы, связанные с выделением вредных паров или газов, должны проводиться в вытяжных шкафах. Проводить работы при неисправной или невключенной вентиляции запрещается.
43.2.4. Необходимо помнить, что дымящие кислоты - соляная, серная, азотная, плавиковая и 25 %-ная аммиачная вода относятся к группе сильнодействующих химических веществ;
Работы по переносу кислот и щелочей более 5 кг осуществляют два человека, переносят бутыль в специальных обрешетках;
в местах хранения азотной кислоты нельзя допускать скопления пыли, соломы и др. воспламеняющихся веществ;
при разбавлении серной кислоты ее следует медленно приливать в воду. Приливание воды в кислоту категорически запрещается;
применять резиновые шланги в качестве сифона при разливе концентрированных кислот запрещается;
нельзя набирать концентрированные кислоты и щелочи в пипетки ртом, только резиновой грушей;
работа с концентрированными кислотами и щелочами без защитных приспособлений (очки, перчатки) запрещается;
концентрированные азотная, серная и соляная кислоты должны храниться в лаборатории в толстостенных банках емкостью не более 2 л, в вытяжном шкафу. Склянки с дымящей азотной кислотой следует хранить в специальных ящиках из нержавеющей стали;
работа с плавиковой кислотой требует особой осторожности;
кислоты, щелочи и другие едкие жидкости следует разливать при помощи стеклянных сифонов грушей или сжатым воздухом, только в вытяжном шкафу;
при случайном разливе кислот, их сначала засыпают песком для впитывания, потом песок убирают и засыпают содой или известью, моют водой и вытирают насухо;
при случайном разливе едкого натра, едкого кали или аммиака можно засыпать песком или опилками, после того как песок уберут смывают слабым раствором уксусной кислоты;
отработанные кислоты и щелочи собирают раздельно в специальную посуду и только после нейтрализации в конце рабочего дня сливают в канализацию или в другое специально отведенное для этой цели место;
слив в канализацию концентрированных кислот и щелочей запрещается.
43.3. Требования пожарной безопасности
43.3.1. Работники лаборатории обязаны знать возможные причины возникновения пожаров и способы их тушения:
запрещается хранить горючие жидкости в вытяжных шкафах при наличии в них горелок или других нагревательных приборов, а также рядом с окислителями (хлоратами, нитратами, азотной кислотой, бромом, перекисью водорода, перманганатами и др.);
вещества, легко отдающие свой кислород (перекись водорода, перекись натрия, магния и др. могут взрываться при взаимодействии с восстановителями;
запрещается работать с горючими жидкостями без включенной вентиляции и курить во время работы;
запрещается сливать горючие жидкости в канализацию, отработанные жидкости собираются в специальную закрывающуюся тару, в конце работы передают на регенерацию или уничтожение.
43.3.2. В случае воспламенения жидкостей (возникновения пожара) необходимо:
отключить вентиляцию, горелки и нагревательные приборы;
вынести из помещения все сосуды с огнеопасными веществами и баллоны со сжатыми газами;
применить наиболее эффективные для данного случая средства пожаротушения.
Пламя тушить нужно прежде всего песком, кошмой, огнетушителем, а где можно - водой.
43.4.8. Меры первой помощи при работе в химической лаборатории:
при термических ожогах первой степени обоженное место надо присыпать двууглекислым натрием, крахмалом, тальком или делать примочки из 2 %-ных растворов питьевой соды или марганцовокислого калия. Лучшим является 96 %-ный этиловый спирт;
при ожогах химическими веществами особенно кислотами и щелочами, пораженный участок быстро промывают большим количеством воды, затем накладывают примочку: при ожогах кислотой - из 2 %-ного раствора соды, при ожогах щелочью из 1 - 2 %-ного раствора уксусной кислоты;
ожоги глаз могут быть не только от попадания в глаза брызг едких веществ и твердых частиц, но и от их аэрозолей и паров. При попадании в глаза паров кислот следует промыть их обильным количеством воды, затем 2 %-ным раствором бикарбоната натрия, все остальное делает врач. Лучшая профилактика ожога глаз - очки.
При воспламенении одежды необходимо загасить огонь на горящем (не бегать!), набросив на него асбестовое или шерстяное одеяло, халат, пальто и т.п. Погасив огонь приступить к оказанию первой помощи;
при порезах рук стеклом необходимо в первую очередь удалить из раны мелкие осколки, промыть 2 %-ным раствором перманганата калия или спиртом, смазать иодной настойкой, перевязать.
ВСЕСОЮЗНЫЙ НАУЧНО-ИССЛЕДОВАТЕЛЬСКИЙ И ПРОЕКТНЫЙ ИНСТИТУТ ТЕХНОЛОГИИ ХИМИЧЕСКОГО И НЕФТЯНОГО АППАРАТОСТРОЕНИЯ
(ВНИИПТхимнефтеппартуры)
|
Директор |
В.А. Самойлов |
|
Заместитель директора |
А.Г. Ламзин |
|
Заведующий отделом стандартизации |
Ю.А. Гук |
|
Заведующий отделом № 29 |
А.П. Окенко |
|
Заведующая лабораторией |
В.В. Раевская |
|
Старший научный сотрудник |
Ю.К. Максимец |
СПИСОК ЛИТЕРАТУРЫ
|
1. |
Гиллебранд В.Ф. Практическое руководство по неорганическому анализу, Госхимиздат, Москва, 1957. |
|
2. |
Дымов A.M. Технический анализ. М., «Металлургия», 1964. |
|
3. |
Степин В.В., Силаева Е.В. и др. Анализ черных металлов, сплавов и марганцевых руд. М., Изд-во черной и цветной металлургии, 1964. |
|
4. |
Теплоухов В.И. Экспресс-анализ стали. М., Изд-во черной и цветной металлургии, 1961. |
|
5. |
Пешкова В.М., Громова М.И. Практическое руководство по спектрофотометрии и колориметрии. М., Изд-во МГУ, 1965. |
|
6. |
Химический и спектральный анализ в металлургии. Практическое руководство. М., «Наука», 1965. |
|
7. |
Конкин В.Д., Клемешов Г.А., Никитина О.И. Методы химического, физикохимического и спектрального анализа сырья, металла и шлака на металлургических заводах. Харьков, Изд-во черной и цветной металлургии, 1961. |
|
8. |
Бабко А.К., Марченко А.В., Фотометрический анализ. Методы определения неметаллов, М., «Химия», 1974. |
|
9. |
Шарло Г., Методы аналитической химии. Количественный анализ неорганических соединений, М., «Химия», 1966. |
|
10. |
Редкоземельные элементы. Изд-во Академии наук СССР, Москва, 1963. |
|
11. |
Сендел Е., Колориметрические методы определения следов металлов, Изд-во «Мир», Москва, 1964. |
|
12. |
Коростелев П.П., Реактивы и растворы в металлургическом анализе. Москва, Изд-во «Металлургия», 1977. |
|
13. |
Редкоземельные элементы. Изд-во Академии наук СССР, Москва, 1963. |
|
14. |
Васильева М.Т., Малыкина В.М. и др. Анализ бора и его соединений, М., Атомиздат, 1965. |
|
15. |
Конкин В.Д., Жихарева В.И. Комплексонометрический анализ, Издательство «Техника», Киев, 1964. |
|
16. |
Еремин Ю.Г., Раевская В.В. и др. «Заводская лаборатория», 1964, № 12. |
|
17. |
Еремин Ю.Г., Раевская В.В., Романов П.Н. Известия высших учебных заведений. «Химия и химическая технология», т. IX, вып. 6, 1966. |
|
18. |
Еремин Ю.Г., Раевская В.В., Романов П.Н. «Журнал аналитической химии», 1966, т. XXI, 11, стр. 1303 |
|
19. |
Еремин Ю.Г., Раевская В.В., Романов П.Н. «Заводская лаборатория», 1962, № 2. |
|
20. |
ГОСТ 12344-66 - ГОСТ 12365-66, ГОСТ 17051-71. Стали (легированные и высоколегированные). Методы химического анализа. |
|
21. |
ГОСТ 12347-77. Стали легированные и высоколегированные. Методы определения фосфора. |
|
22. |
ГОСТ 12344-78, ГОСТ 12346-78, ГОСТ 12348-78, ГОСТ 12350-78, ГОСТ 12353-78, ГОСТ 12355-78. Стали легированные и высоколегированные. Методы химического анализа. |
|
23. |
ГОСТ 22536.0-77 - ГОСТ 22536.13-77. Сталь углеродистая и чугун нелегированный. Методы анализа. |
|
24. |
Мандельштам С.Л. Введение в спектральный анализ. Гостехиздат, 1946. |
|
25. |
Свентицкий Н.С. Визуальные методы эмиссионного спектрального анализа, М., 1961. |
|
26. |
Куделя Е.С. «Заводская лаборатория», 1951, № 1, 1952, № 4. |
|
27. |
Зимина А.А. «Заводская лаборатория», 1953, № 1. |
|
28. |
Белькевич Я.П. Полуколичественный анализ сталей на стилоскопе. Судпромгиз, 1957. |
|
29. |
Никифорова Е.Ф. руководство по спектральному анализу черных металлов на стилоскопе. Трансжелдориздат, 1950. |
|
30. |
Свентицкий Н.С. Стилоскоп. Гостеоретиздат, 1948. |
|
31. |
Ломоносова Л.С., Фалькова О.Б. Спектральный анализ. М., Металлургиздат, 1958. |
|
32. |
Бураков B.C., Янковский А.А. Практическое руководство по спектральному анализу. Изд-во АН БССР, 1960. |
|
33. |
Кустанович И.М. Спектральный анализ. М., Госиздат, 1962. |
|
34. |
Фотоэлектрические методы спектрального анализа. Сб., Оборонгиз, 1961. |
|
35. |
Фишман И.С. Методы количественного спектрального анализа. Казань, Изд-во Казанского университета, 1962. |
|
36. |
Славный В.А., Абрамсон И.С. Материалы семинара по проблеме повышения точности, чувствительности и правильности спектрального, анализа. Сб., т. 1. МДНТИ, 1964. |
|
37. |
Шаевич А.Б. Некоторые вопросы эмиссионной и молекулярной спектроскопии. М., Металлургиздат, 1960. |
|
38. |
Нагибина И.М., Прокофьев В.К. Спектральные приборы и техника спектроскопии. Л., Машгиз, 1963. |
|
39. |
Охрана труда в научных учреждениях Академии Наук СССР. Наука, 1972. |
|
40. |
Сборник типовых инструкций по охране труда. Москва, Недра, 1978. |
Приложение
Текущие, капитальные и приведенные затраты на выполнение одного анализа (химическими методами)
Примечания к приложению
текущие затраты на выполнение одного анализа складываются из суммы зарплаты лаборантов, амортизации на оборудование, занятого при выполнении анализа и стоимости химических реактивов, применяемых для одного анализа;
капитальные вложения включают в себя стоимость оборудования, относимого на выполнение одного анализа;
приведенные затраты включают в себя текущие затраты и капиталовложения, умноженные на нормативный коэффициент, равный 0,15.
СОДЕРЖАНИЕ