 | Информационная система | |
4.1. МЕТОДЫ КОНТРОЛЯ. ХИМИЧЕСКИЕ ФАКТОРЫ
Определение остаточных количеств
пестицидов в пищевых продуктах,
сельскохозяйственном сырье и объектах
окружающей среды
Сборник
МУК 4.1.2859 - 4.1.2866-11
1. Разработаны Российским государственным аграрным университетом - МСХА им. К.А. Тимирязева, Учебно-научный консультационный центр «Агроэкология пестицидов и агрохимикатов» Минсельхоза России (В.А. Калинин, профессор, канд. с-х. наук., Е.В. Довгилевич, ст.н.сотр., канд. биол. наук, А.В. Довгилевич, ст.н.сотр., канд. хим. наук, Н.В. Устименко, ст.н.сотр., канд. биол. наук, Е.Н. Щербинкина, инженер).
2. Рекомендованы к утверждению Комиссией по санитарно-эпидемиологическому нормированию при Федеральной службе по надзору в сфере защиты прав потребителей и благополучия человека (протокол от 28 12.2010 № 3).
3. Утверждены руководителем Федеральной службы по надзору в сфере защиты прав потребителей и благополучия человека, Главным государственным санитарным врачом Российской Федерации Г.Г. Онищенко 31 марта 2011 г.
4. Введены в действие с момента утверждения.
СОДЕРЖАНИЕ
|
|
УТВЕРЖДАЮ Руководитель
Федеральной службы Г.Г. Онищенко 31 марта 2011 г. Дата введения: с момента утверждения |
4.1. МЕТОДЫ КОНТРОЛЯ. ХИМИЧЕСКИЕ ФАКТОРЫ
Методика
измерений остаточных количеств
Тиабендазола в семенах и масле рапса методом
высокоэффективной жидкостной хроматографии
Методические указания
МУК 4.1.2864-11
Общие положения и область применения
Свидетельство об аттестации методики от 23.11.2010 № 01.5.04.693.
Настоящие методические указания устанавливают метод измерения массовой концентрации Тиабендазола в семенах и масле рапса в диапазоне концентраций 0,01 - 0,1 мг/кг.
1. Краткая характеристика препарата
Действующее вещество: Тиабендазол.
Структурная формула:
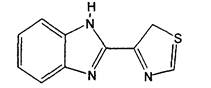
Название действующего вещества по ИЮПАК: 2-(1,3-тиазол-4-ил) бензимидазол.
2-(4-тиазолил)-1Н-бензимидазол (С.А.).
Эмпирическая формула: C10H7N3S.
Белое кристаллическое вещество. Температура плавления: 297 - 298 °С. Давление паров при 25 °С: 4,6×10-4 мПа. Коэффициент распределения н-октанол-вода: Kow logР = 2,39 (рН 7).
Растворимость (г/л) при 20 °С: н-гептан - < 0,01, ксилол - 0,13, ацетон - 2,43, 1,2-дихлорэтан - 0,81, метанол - 8,28, этилацетат - 1,49, н-октанол - 3,91, вода - 10,0 (рН 2), 0,16 (рН 4), 0,03 (рН 7 - 10). Устойчив к гидролизу. Водный фотолиз (20 °С, рН 5, DT50 29 ч); pКa1 = 4,73; рКа2 = 12,00.
Краткая токсикологическая характеристика: Острая пероральная токсичность (LD50) для мышей - 3600 мг/кг, крыс - 3100 мг/кг, кроликов - 3800 мг/кг, птиц - более 2250 мг/кг; острая дермальная токсичность (LD50) для кроликов - более 2000 мг/кг, не ирритант; ингаляционная токсичность (LС50) для крыс - более 0,5 мг/кг. Не токсичен для пчел. СК50 для рыб 0,55 мг/л при экспозиции 96 ч. Класс токсичности по ВОЗ-III.
Область применения: Тиабендазол - системный фунгицид защитного и лечебного действия. Тиабендазол образует защитный слой на обработанной поверхности, используется на овощных, плодовых и зерновых культурах. Не совместим с купратами и окисляющими агентами, такими как хлораты и нитраты.
Гигиенические нормативы для Тиабендазола в семенах и масле рапса в России не установлены.
2. Методика определения Тиабендазола в семенах и масле рапса методом ВЭЖХ
2.1. Основные положения
2.1.1. Принцип метода
Методика основана на определении Тиабендазола методом ВЭЖХ с использованием УФ-детектора после его извлечения из образцов органическим растворителем с последующей очисткой на колонке с силикагелем.
2.1.2. Метрологическая характеристика метода
При соблюдении всех регламентированных условий проведения анализа в точном соответствии с данной методикой погрешность (и ее составляющие) результатов измерений при доверительной вероятности Р = 0,95 не превышает значений, приведенных в табл. 1 для соответствующих диапазонов концентраций.
Метрологические параметры
|
Диапазон измерений, массовая концентрация, мг/кг |
Показатель повторяемости (относительное среднеквадратическое отклонение повторяемости), σr % |
Показатель промежуточной прецизионности (относительное среднеквадратическое отклонение в условиях вариации факторов «время», «оператор» в одной лаборатории), σRл, % |
Показатель воспроизводимости (относительное среднеквадратическое отклонение воспроизводимости), σR, % |
Показатель точности* (границы относительной погрешности при вероятности Р = 0,95), ±δ, % |
|
Семена рапса от 0,01 до 0,10 вкл. |
5 |
12 |
14 |
24 |
|
Масло рапса от 0,01 до 0,10 вкл. |
6 |
7 |
8 |
16 |
|
* Соответствует расширенной неопределенности Uотн при коэффициенте охвата k = 2. |
||||
Полнота извлечения вещества, стандартное отклонение, доверительный интервал среднего результата для полного диапазона концентраций (n = 20) приведены в табл. 2.
|
Объект анализа |
Предел обнаружения, мг/кг |
Диапазон измеряемых концентраций, мг/кг |
Среднее значение определения, % |
Стандартное отклонение, S, % |
Доверительный интервал среднего результата, ±% |
|
Семена рапса |
0,01 |
0,01 - 0,1 |
89,4 |
2,7 |
5,3 |
|
Масло рапса |
0,01 |
0,01 - 0,1 |
89,8 |
1,1 |
2,3 |
2.1.3. Избирательность метода
Избирательность метода определения Тиабендазола достигается сочетанием условий подготовки проб и хроматографического анализа.
2.2. Реактивы и материалы
|
Ацетон, осч |
ТУ 6-09-3513-86 |
|
Ацетонитрил для ВЭЖХ, сорт 1 или хч |
ТУ 6-09-3534-87 |
|
Бумажные фильтры «красная лента» |
ТУ 2642-001-42624157-98 |
|
Вода бидистиллированная, деионизированная |
ГОСТ 6709-79 |
|
н-Гексан, хч, свежеперегнанный |
ТУ 2631-003-05807999-98 |
|
Изопропиловый спирт |
ТУ 2632-015-11291058-95 |
|
Калий углекислый, хч |
|
|
Калия перманганат |
|
|
Кальция хлорид, хч |
ГОСТ 4161-77 |
|
Кислота ортофосфорная, хч, 0,005 М водный раствор |
|
|
Кислота серная, хч |
|
|
Натрий двууглекислый |
|
|
Натрий сернокислый безводный, ч, свежепрокаленный |
|
|
Натрий хлористый, чда |
|
|
Натрия гидроксид, хч |
|
|
Подвижная фаза для ВЭЖХ: ацетонитрил: 0,005 М раствор Н3РО4 (40:60, по объему) |
|
|
Силикагель для колоночной хроматографии 60 (0,040 - 0,063 mm) («Merck», Германия) |
|
|
Стекловата |
|
|
Тиабендазол, аналитический стандарт с содержанием д.в. 99,8 % (Cheminova) |
|
|
Элюент № 1 для колоночной хроматографии: смесь гексан - ацетон (80:20, по объему) |
|
|
Элюент № 2 для колоночной хроматографии: смесь гексан - ацетон (15:85, по объему) |
|
Допускается использование реактивов квалификацией не ниже указанных.
2.3. Приборы и посуда
|
Жидкостный хроматограф «Альянс» фирмы «Waters» с УФ-детектором (Waters 2487) с дегазатором и автоматическим пробоотборником или аналогичный |
|
|
Колонка Spherisorb Phenyl (150×4,6) мм, 3 мкм (Waters, USA) или аналогичная |
|
|
Ванна ультразвуковая УЭВ-1,3, или аналогичная |
|
|
Весы аналитические BЛA-200 или аналогичные |
|
|
Мельница лабораторная зерновая ЛMЗ |
ТУ 1-01-0593-79 |
|
Гомогенизатор |
МРТУ 42-1505-63 |
|
Ротационный испаритель вакуумный Buchi R-205 или аналогичный |
|
|
Бидистиллятор рН-метр универсальный ЭВ-74 |
ГОСТ 22261-76 |
|
Насос водоструйный |
МРТУ 42 861-64 |
|
Колбы плоскодонные на шлифах КШ500 29/32 ТС |
ГОСТ 10384-72 |
|
Колбы круглодонные на шлифах КШ10 и КШ250 29-32 ТС |
ГОСТ 10384-72 |
|
Воронки лабораторные В-75-110 |
|
|
Воронки делительные ВД-3-250 и 500 |
ГОСТ 8613-75 |
|
Цилиндры мерные на 50, 100, 500 и 1000 см3 |
ГОСТ 1774-74 |
|
Колбы мерные на 25, 50, 100 и 1000 см3 |
|
|
Пипетки на 1, 2, 5, 10 см3 |
ГОСТ 22292-74 |
|
Колонки стеклянные (25×1) см |
|
Допускается использование приборов и посуды с метрологическими характеристиками не ниже указанных.
2.4. Отбор проб
Отбор проб семян рапса производится в соответствии с ГОСТ 10852-86 «Семена масличные. Правила приемки и методы отбора проб». Семена хранят при комнатной температуре в полотняных мешочках, перед анализом доводят до стандартной влажности и измельчают. Растительное масло хранят в холодильнике при температуре 0 - 4 °С в герметично закрытой стеклянной таре в течение 2 месяцев.
2.5. Подготовка к определению
2.5.1. Подготовка и очистка реактивов и растворителей
Перед началом работы проверяют чистоту применяемых органических растворителей. Для этого 100 см3 растворителя упаривают в ротационном вакуумном испарителе при температуре 40 °С до объема 1,0 см3 и хроматографируют. При обнаружении мешающих определению примесей очистку растворителей производят в соответствии с общепринятыми методиками.
2.5.2. Кондиционирование колонки
Перед началом анализа колонку (Spherisorb Phenyl) кондиционируют в потоке подвижной фазы (1 см3/мин) до стабилизации нулевой линии в течение 1 - 2 ч.
2.5.3. Приготовление растворов
Для приготовления 0,005 М раствора ортофосфорной кислоты 0,5 г 98 % ортофосфорной кислоты помещают в мерную колбу объемом 1 дм3, растворяют в бидистиллированной воде и доводят объем до метки.
Для приготовления подвижной фазы смешивают ацетонитрил с 0,005 М раствором ортофосфорной кислоты в соотношении 50:50 по объему, используя мерные цилиндры.
Для приготовления элюента № 1 в колбе на 1000 мл смешивают 800 мл н-гексана и 200 мл ацетона. Для приготовления элюента № 2 в колбе на 1000 мл смешивают 150 мл н-гексана и 850 мл ацетона.
Приготовление стандартного и градуировочных растворов:
Берут точную навеску Тиабендазола (50 мг), переносят в мерную колбу на 50 мл, растворяют навеску в ацетонитриле и доводят до метки (стандартный раствор с концентрацией 1,0 мг/см3).
Градуировочные растворы с концентрациями 0,1, 0,2, 0,5, 1,0 и 2,0 мгк/cм3 готовят методом последовательного разбавления по объему, используя раствор подвижной фазы [смесь ацетонитрил - 0,005 М Н3РО4 (40:60, по объему)]. Стандартный и градуировочные растворы можно хранить в холодильнике при температуре 0 - 4 °С в течение 1 месяца.
Для внесения в контрольный образец при определении полноты извлечения используют стандартный раствор, разбавленный ацетонитрилом до нужного уровня концентраций методом последовательного разбавления. Растворы для внесения в масло готовят из стандартного раствора с концентрацией 0,5 мг/см3 методом последовательного разбавления по объему изопропиловым спиртом.
2.5.4. Построение градуировочного графика
Для построения градуировочного графика (площадь пика - концентрация Тиабендазола в растворе) в хроматограф вводят по 20 мм3 градуировочных растворов (не менее 3 параллельных измерений для каждой концентрации, не менее 4 точек по диапазону измеряемых концентраций), измеряют площади пиков и строят график зависимости среднего значения площади пика от концентрации Тиабендазола в градуировочном растворе (мкг/см3).
Градуировочную характеристику необходимо проверять при замене реактивов, хроматографической колонки или элементов хроматографической системы, а также при отрицательном результате контроля градуировочного коэффициента:
![]()
λконтр. - норматив контроля градуировочного коэффициента, %. (λконтр. = 10 % при Р = 0,95).
2.5.5. Подготовка колонки с силикагелем для очистки экстракта
В нижнюю часть стеклянной колонки длиной 25 см и внутренним диаметром 1 см помещают тампон из стекловаты, закрывают кран и вносят суспензию 5 г силикагеля в 30 см3 смеси гексан-ацетон (80:20, по объему). Дают растворителю стечь до верхнего края сорбента. Колонку последовательно промывают 30 см3 элюента № 2 и 30 см3 элюента № 1 со скоростью 1 - 2 капли в секунду, после чего она готова к работе.
2.5.6. Проверка хроматографического поведения Тиабендазола на колонке с силикагелем
В круглодонную колбу емкостью 10 см3 отбирают 0,1 см3 стандартного раствора Тиабендазола с концентрацией 10 мкг/см3. Отдувают растворитель током теплого воздуха (температура не выше 40 °С), остаток растворяют в 5 см3 элюента № 1 и наносят на колонку. Колбу обмывают еще 5 см3 элюента № 1 и также вносят на колонку. Промывают колонку 50 см3 элюента № 1, которые отбрасывают, затем 100 см3 элюента № 2 со скоростью 1 - 2 капли в секунду. Отбирают фракции по 10 см3 каждая, упаривают, остаток растворяют в 2 см3 подвижной фазы для ВЭЖХ (п. 2.5.3) и анализируют на содержание Тиабендазола (п. 2.6.3).
Фракции, содержащие Тиабендазол, объединяют, упаривают досуха, остаток растворяют в 2 см3 подвижной фазы для ВЭЖХ и вновь анализируют (п. 2.6.3). Рассчитывают содержание Тиабендазола в элюате, определяя полноту вымывания вещества из колонки и необходимый для этого объем элюента.
Примечание: параметры удерживания Тиабендазола и сопутствующих экстрактивных веществ могут меняться при использовании новой партии сорбента и растворителей.
2.5.7. Подготовка приборов и средств измерения
Установка и подготовка всех приборов и средств измерения проводится в соответствии с требованиями технической документации.
2.6. Проведение определения
2.6.1. Извлечение Тиабендазола из проб семян и масла рапса
Навеску измельченных семян массой 20 г помещают в коническую колбу емкостью 100 мл и экстрагируют Тиабендазол 40 см3 ацетонитрила на ультразвуковой установке в течение 15 мин. Суспензию фильтруют через бумажный фильтр «красная лента». Экстракцию повторяют дважды порциями по 30 см3.
Навеску масла массой 20 г помещают в делительную воронку емкостью 250 см3 и экстрагируют Тиабендазол трижды ацетонитрилом порциями по 30 см3, встряхивая смесь каждый раз в течение 2 - 3 мин и собирая нижний органический слой.
Объединенные экстракты семян или масла промывают гексаном в делительных воронках дважды по 25 см3, встряхивая смесь каждый раз в течение 1 - 2 мин и собирая нижний органический слой.
После этого экстракты выпаривают досуха на ротационном испарителе при температуре не выше 40 °С. Дальнейшую очистку экстрактов проводят на колонке с силикагелем по п. 2.6.2.
Внимание! Отделение ацетонитрильного слоя следует производить только после полного расслоения жидкостей в делительной воронке.
2.6.2. Очистка на колонке с силикагелем
Сухой остаток в колбе, полученный при упаривании очищенных по п. 2.6.1 экстрактов растительных материалов, количественно переносят двумя порциями смеси гексан-ацетон (80:20, по объему) по 5 см3 каждая в кондиционированную хроматографическую колонку (п. 2.5.5). Промывают колонку 50 см3 элюента № 1, который отбрасывают. Тиабендазол элюируют 80 см3 элюента № 2, собирая элюат в грушевидную колбу емкостью 250 см3. Раствор упаривают досуха на вакуумном ротационном испарителе при температуре не выше 40 °С. Сухой остаток растворяют в 2 см3 подвижной фазы для ВЭЖХ и 20 мм3 раствора вводят в жидкостный хроматограф.
2.6.3. Условия хроматографирования
Жидкостный хроматограф «Альянс» фирмы «Waters» с УФ-детектором (Waters 2 487), снабженный дегазатором, автоматическим пробоотборником и термостатом колонки.
Колонка Spherisorb Phenyl (150×4,6) мм, 3 мкм (Waters, USA).
Температура колонки 30 ± 1 °С.
Подвижная фаза для ВЭЖХ: ацетонитрил: 0,005 М раствор Н3РО4 (40:60, по объему).
Скорость потока элюента: 1 мл/мин.
Рабочая длина волны 300 нм.
Объем вводимой пробы 20 мм3.
Линейный диапазон детектирования 0,1 - 2,00 мкг/мл.
2.6.4. Обработка результатов анализа
Количественное определение проводят методом абсолютной калибровки, содержание Тиабендазола в образцах семян или масла (X, мг/кг) вычисляют по формуле:
![]()
S1 - площадь пика Тиабендазола в стандартном растворе, мм2 (мв∙сек);
S2 - площадь пика Тиабендазола в анализируемой пробе, мм2 (мв∙сек);
V - объем пробы, подготовленной для хроматографического анализа, см3;
Р - навеска анализируемого образца, г;
С - концентрация стандартного раствора Тиабендазола, мкг/см3;
Содержание остаточных количеств Тиабендазола в анализируемом образце вычисляют как среднее из 2 параллельных определений.
Образцы, дающие пики большие, чем стандартный раствор Тиабендазола 2 мкг/см3, разбавляют подвижной фазой для ВЭЖХ.
3. Проверка приемлемости результатов параллельных определений
За результат анализа принимают среднее арифметическое результатов двух параллельных определений, расхождение между которыми не превышает придела повторяемости (1):
X1, Х2 - результаты параллельных определений, мг/кг;
r - значение предела повторяемости (r = 2,8σr).
При невыполнении условия (1) выясняют причины превышения предела повторяемости, устраняют их и вновь выполняют анализ.
4. Оформление результатов
Результат анализа представляют в виде:
(X ± Δ) мг/кг при вероятности Р = 0,95, где
X - среднее арифметическое результатов определений, признанных приемлемыми, мг/кг;
Δ - граница абсолютной погрешности, мг/кг;
Δ = δ∙X/100, где
δ - граница относительной погрешности методики (показатель точности в соответствии с диапазоном концентраций), %.
В случае если содержание компонента менее нижней границы диапазона определяемых концентраций, результат анализа представляют в виде:
«содержание вещества в пробе менее нижней границы определения» (например: менее 0,01 мг/кг*, где * - 0,01 мг/кг - предел обнаружения Тиабендазола в рапсе).
5. Контроль качества результатов измерений
Оперативный контроль погрешности и воспроизводимости измерений осуществляется в соответствии с ГОСТ Р ИСО 5725-1-6-2002 «Точность (правильность и прецизионность) методов и результатов измерений».
5.1. Стабильность результатов измерений контролируют перед проведением измерений, анализируя один из градуировочных растворов.
5.2. Плановый внутрилабораторный оперативный контроль процедуры выполнения анализа проводится с применением метода добавок.
Величина добавки Cд должна удовлетворять условию:
Cд = Δл,Х + Δл,Х', где
±Δл,Х (±Δл,Х') - характеристика погрешности (абсолютная погрешность) результатов анализа, соответствующая содержанию компонента в испытуемом образце (расчетному значению содержания компонента в образце с добавкой, соответственно), мг/кг; при этом:
Δл = ±0,84Δ, где
Δ - граница абсолютной погрешности, мг/кг: где
Δ = δ∙X/100, где
δ - граница относительной погрешности методики (показатель точности в соответствии с диапазоном концентраций), %.
Результат контроля процедуры Кк рассчитывают по формуле:
Кк = X' - X - Cд, где
X', X, Cд - среднее арифметическое результатов параллельных определений (признанных приемлемыми по п. 4) содержания компонента в образце с добавкой, испытуемом образце, концентрация добавки соответственно, мг/кг.
Норматив контроля К рассчитывают по формуле:
![]()
Проводят сопоставление результата контроля процедуры (Кк) с нормативом контроля (К).
Если результат контроля процедуры удовлетворяет условию:
|
|Кк| ≤ К, |
(2) |
процедуру анализа признают удовлетворительной.
При невыполнении условия (2) процедуру контроля повторяют. При повторном невыполнении условия (2) выясняют причины, приводящие к неудовлетворительным результатам, и принимают меры к их устранению.
5.3. Проверка приемлемости результатов измерений, полученных в условиях воспроизводимости:
Расхождение между результатами измерений, выполненных в двух разных лабораториях, не должно превышать предела воспроизводимости (R):
|
|
(3) |
Х1, Х2 - результаты измерений в двух разных лабораториях, мг/кг;
R - предел воспроизводимости (в соответствии с диапазоном концентраций), %.
6. Требования безопасности
6.1. При работе необходимо соблюдать требования техники безопасности, установленные для работ с токсичными, едкими, легковоспламеняющимися веществами (ГОСТ 12.1.005, ГОСТ 12.1.007). Организация обучения работников безопасности труда по ГОСТ 12.0.004.
При выполнении измерений с использованием жидкостного хроматографа и работе с электроустановками соблюдать правила электробезопасности в соответствии с ГОСТ 12.1.019-79 и инструкциями по эксплуатации приборов.
6.2. Помещение лаборатории должно быть оборудовано приточно-вытяжной вентиляцией, соответствовать требованиям пожарной безопасности по ГОСТ 12.1.004-91 и иметь средства пожаротушения по ГОСТ 12.4.009. Содержание вредных веществ в воздухе не должно превышать норм, установленных ГН 1.2.2701-10 и ГН 2.2.5.1313-03 «Предельно допустимые концентрации (ПДК) вредных веществ в воздухе рабочей зоны».
7. Требования к квалификации оператора
Измерения в соответствии с настоящей методикой может выполнять специалист-химик, имеющий опыт работы методом высокоэффективной жидкостной хроматографии, ознакомленный с руководством по эксплуатации жидкостного хроматографа, освоивший данную методику и подтвердивший экспериментально соответствие получаемых результатов нормативам контроля погрешности измерений по п. 5.
8. Разработчики
Цибульская И.А., Черменская Т.Д., Ковров Н.Г. (ГНУ Всероссийский научно-исследовательский институт защиты растений Россельхозакадемии, г. Санкт-Петербург).
Методика прошла метрологическую экспертизу (Свидетельство об аттестации № 01.5.04.686) и внесена в Федеральный реестр (ФР. 1.31.2011.09107).