 | Информационная система | |
ГОСТ 29117-91
МЕЖГОСУДАРСТВЕННЫЙ СТАНДАРТ
СТАЛИ ЛЕГИРОВАННЫЕ
И ВЫСОКОЛЕГИРОВАННЫЕ
МЕТОД ОПРЕДЕЛЕНИЯ ВИСМУТА
ИПК
ИЗДАТЕЛЬСТВО СТАНДАРТОВ
Москва
МЕЖГОСУДАРСТВЕННЫЙ СТАНДАРТ
|
СТАЛИ ЛЕГИРОВАННЫЕ И ВЫСОКОЛЕГИРОВАННЫЕ Методы определения висмута Alloyed and
high-alloyed steels. |
ГОСТ |
Дата введения 01.01.93
Настоящий стандарт устанавливает фотометрический (при массовых долях от 0,0005 % до 0,01 %), непламенный атомно-абсорбционный (при массовых долях от 0,0001 % до 0,01 %), инверсионно-вольтамперометрический (при массовых долях от 0,0001 % до 0,005 %) и полярографический (при массовых долях от 0,001 % до 0,01 %) методы определения висмута в легированных и высоколегированных сталях.
1. ОБЩИЕ ТРЕБОВАНИЯ
Общие требования к методам анализа - по ГОСТ 28473.
2. ФОТОМЕТРИЧЕСКИЙ МЕТОД
2.1. Сущность метода
Метод основан на образовании окрашенного в розовый цвет комплексного соединения висмута с ксиленоловым оранжевым в азотной кислоте концентрацией 0,1 моль/дм3 и измерении его оптической плотности в области светопропускания с максимумом поглощения при длине волны 540 нм.
Висмут предварительно отделяют от сопутствующих элементов стали осаждением в виде сульфида тиоацетамидом в аммиачном растворе в присутствии коллектора сульфида меди и винной кислоты в качестве комплексообразующего вещества или методом ионообменной хроматографии.
2.2. Аппаратура, реактивы и растворы
Спектрофотометр или фотоэлектроколориметр со всеми принадлежностями для измерения в видимой области спектра.
pH-метр.
Термометр со шкалой до 150 °C.
Ионообменная колонка диаметром 1,2 - 1,5 см, высотой 30 - 40 см, заполненная анионитом АН-31, АВ-17-8 или АВ-17-8чС в Cl--форме с высотой слоя 15 см.
Анионит АН-31 по ГОСТ 20301.
Подготовка анионита к анализу: 200 г анионита заливают не менее чем пятикратным объемом насыщенного раствора хлористого натрия и оставляют для набухания на 24 ч.
Для отделения фракции смолы с размером зерна менее 0,4 мм взвесь смолы в растворе хлористого натрия выливают на сито с сеткой № 063 по ГОСТ 6613 и промывают струей воды, собирая прошедшую сквозь сито фракцию анионита вместе с водой в сосуд вместимостью 2 - 3 дм3. Оставшуюся на сите смолу отбрасывают. Жидкость над фракцией анионита, прошедшей сквозь сито, декантируют, а анионит промывают способом декантации соляной кислотой концентрацией вначале 3 моль/дм3, затем 0,5 моль/дм3 до полного удаления Fe (III) (проба с роданистым калием). Анионит промывают 10-кратным количеством воды, затем заливают на 48 ч раствором гидроксида натрия. Далее анионит промывают водой до нейтральной реакции по универсальному индикатору и хранят под водой в стеклянной банке с притертой пробкой. Перед началом работы в нижнюю часть хроматографической колонки помещают тампон из полихлорвиниловой нити или из стеклянной ваты, предварительно прокипяченной в соляной кислоте (1:1) и промытой водой. Ионообменную колонку заполняют анионитом на высоту слоя 15 см и затем водой на 3/4 ее высоты. Слой анионита должен быть ровным, без пузырьков воздуха. Для переведения смолы в Cl--форму через нее пропускают 100 - 150 см3 соляной кислоты концентрацией 3 моль/дм3 со скоростью 1 см3/мин.
Анионит АВ-17-8 или АВ-17-8чС по ГОСТ 20301.
Подготовка анионита к анализу: 200 см3 товарного анионита АВ-17-8 или АВ-17-8чС (выпускаемого в виде взвеси в воде) промывают дважды водой способом декантации. Для отделения фракции смолы с размером зерна менее 0,6 мм взвесь смолы в воде выливают на сито с сеткой № 063 по ГОСТ 6613 и промывают струей воды, собирая прошедшую сквозь сито фракцию анионита вместе с водой в сосуд вместимостью 2 - 3 дм3. Оставшуюся на сите смолу отбрасывают. Фракцию анионита, прошедшую сквозь сито, подвергают подготовке к анализу, как смолу АН-31.
Кислота соляная по ГОСТ 3118 или ГОСТ 14261, разбавленная 1:1, и растворы концентрацией 3 моль/дм3 и 0,5 моль/дм3.
Кислота азотная по ГОСТ 4461 или ГОСТ 11125, разбавленная 1:1, 1:15, 1:500, и растворы концентрацией 1 моль/дм3 и 0,1 моль/дм3.
Смесь соляной и азотной кислот: к 150 см3 соляной кислоты добавляют 50 см3 азотной кислоты, 200 см3 воды и перемешивают. Смесь готовят непосредственно перед использованием.
Кислота аскорбиновая, раствор концентрацией 100 г/дм3, свежеприготовленный.
Кислота винная по ГОСТ 5817, растворы концентрацией 500 г/дм3 и 100 г/дм3.
Аммиак водный по ГОСТ 3760 или ГОСТ 24147 и разбавленный 1:200.
Тиоацетамид, раствор концентрацией 20 г/дм3.
Ксиленоловый оранжевый, раствор концентрацией 1 г/дм3 в азотной кислоте концентрацией 0,1 моль/дм3.
Натрий хлористый по ГОСТ 4233, насыщенный раствор.
Натрия гидроксид по ГОСТ 4328, раствор концентрацией 50 г/дм3.
Калий роданистый по ГОСТ 4139, раствор концентрацией 50 г/дм3.
Медь марки М00б по ГОСТ 859.
Медь азотнокислая, раствор концентрацией 0,01 г/см3: 1 г меди растворяют при нагревании в 15 - 20 см3 азотной кислоты (1:1). Раствор кипятят для удаления оксидов азота, охлаждают, разбавляют водой до 100 см3 и перемешивают.
Железо карбонильное радиотехническое марки Пс по ГОСТ 13610.
Универсальная индикаторная бумага pH 1 - 10.
Висмут марок Ви00 по ГОСТ 10928, марок Ви000 и Ви0000 по ТУ 48-6-11.
Стандартные растворы висмута.
Раствор А: 0,1 г висмута растворяют при нагревании в 30 см3 азотной кислоты, кипятят раствор до удаления оксидов азота, охлаждают, переносят в мерную колбу вместимостью 1 дм3, доливают до метки водой и перемешивают.
1 см3 стандартного раствора А содержит 0,0001 г висмута.
Раствор Б: 10 см3 раствора А помещают в мерную колбу вместимостью 100 см3, добавляют 10 см3 азотной кислоты, доливают до метки водой и перемешивают.
1 см3 стандартного раствора Б содержит 0,00001 г висмута.
2.3. Проведение анализа
2.3.1. Приготовление испытуемого раствора
2.3.1.1. Для сталей, содержащих вольфрам, молибден, ниобий
Навеску стали массой 1 г (при массовых долях висмута от 0,0005 % до 0,002 %) или 0,5 г (при массовых долях висмута от 0,002 % до 0,01 %) помещают в стакан (или колбу) вместимостью 250 - 300 см3, приливают 15 - 20 см3 соляной кислоты, 5 см3 азотной кислоты, накрывают часовым стеклом и растворяют навеску при нагревании.
Раствор выпаривают до объема приблизительно 10 см3, добавляют 30 см3 воды, 20 - 30 см3 раствора винной кислоты концентрацией 500 г/дм3 и нагревают в течение 10 мин. Раствор охлаждают, добавляют 20 - 25 см3 аммиака до pH 8 - 10 по универсальному индикатору и вновь нагревают в течение 10 мин до растворения выделившихся вольфрамовой и молибденовой кислот. Добавляют 1 см3 раствора азотнокислой меди, устанавливают pH 7,5, добавляя соляную кислоту (1:1) или аммиак, используя pH-метр.
Раствор разбавляют водой до 150 см3, нагревают до 85 °C - 90 °C, приливают 10 см3 раствора тиоацетамида, выдерживают 10 мин при этой же температуре и вновь приливают 10 см3 раствора тиоацетамида. Оставляют раствор с осадком на 2 ч при 40 °C - 50 °C.
Затем раствор с осадком охлаждают до комнатной температуры, отфильтровывают осадок сульфидов на два фильтра средней плотности (белая лента), промывают 7 - 8 раз холодной водой, фильтрат отбрасывают. Осадок на фильтре растворяют в 30 - 40 см3 (порциями по 10 см3) горячей смеси соляной и азотной кислот и промывают фильтр 2 - 3 раза горячей азотной кислотой (1:500), собирая фильтрат и промывные воды в стакан, в котором проводилось осаждение. Фильтр отбрасывают.
2.3.1.2. Для сталей, не содержащих вольфрам, молибден, ниобий
Навеску стали массой 1 г (при массовых долях висмута от 0,0005 % до 0,002 %) или 0,5 г (при массовых долях висмута от 0,002 % до 0,01 %) помещают в стакан (или колбу) вместимостью 250 - 300 см3, приливают 15 - 20 см3 соляной кислоты, 5 см3 азотной кислоты, накрывают стакан (или колбу) часовым стеклом и растворяют навеску при нагревании. Раствор выпаривают до влажных солей. Соли растворяют в 5 - 7 см3 соляной кислоты и снова выпаривают до влажных солей. Эту операцию повторяют. Соли растворяют в 40 см3 соляной кислоты концентрацией 3 моль/дм3, нагревая до начала кипения. Образовавшийся осадок кремниевой кислоты отфильтровывают на два фильтра средней плотности (белая лента) и промывают 3 - 4 раза горячей соляной кислотой концентрацией 3 моль/дм3, присоединяя промывную жидкость к основному фильтрату. Фильтр с осадком отбрасывают.
Фильтрат пропускают через ионообменную колонку с анионитом, предварительно промытую 50 см3 соляной кислоты концентрацией 3 моль/дм3 со скоростью приблизительно 0,5 см3/мин. После того, как весь испытуемый раствор перенесен в ионообменную колонку, пропускают еще 70 - 100 см3 (порциями по 10 - 20 см3) соляной кислоты концентрацией 3 моль/дм3 для удаления сопутствующих элементов: никеля, хрома, кобальта, марганца, меди, железа. Элюат отбрасывают. Когда последняя порция кислоты достигнет верхнего уровня анионита, десорбируют висмут 300 см3 азотной кислоты концентрацией 1 моль/дм3.
2.3.2. Спектрофотометрическая процедура анализа
Испытуемый раствор, приготовленный по п. 2.3.1.1 или 2.3.1.2, выпаривают до влажных солей, соли растворяют в 5 см3 азотной кислоты и выпаривают досуха.
Затем соли растворяют в 3 см3 азотной кислоты концентрацией 1 моль/дм3 при нагревании, стенки стакана ополаскивают 3 - 5 см3 воды, раствор перемешивают и охлаждают. Добавляют 2 см3 раствора аскорбиновой кислоты, перемешивают, через 5 мин приливают 1 см3 раствора винной кислоты концентрацией 100 г/дм3 и 1 см3 раствора ксиленолового оранжевого, перемешивая раствор после добавления каждого реактива. Раствор переносят в мерную колбу вместимостью 25 см3, доливают до метки водой и перемешивают.
Через 10 мин измеряют оптическую плотность окрашенного раствора на спектрофотометре при длине волны 540 нм в кювете с толщиной поглощающего свет слоя 1 см или на фотоэлектроколориметре с зеленым светофильтром в кювете с толщиной поглощающего свет слоя 5 см. Раствором сравнения служит раствор контрольного опыта. Массу висмута в испытуемом растворе находят по градуировочному графику.
2.3.3. Построение градуировочного графика
В пять стаканов (или колб) вместимостью 250 - 300 см3 помещают навески карбонильного железа 0,5 - 1 г в соответствии с массой навески анализируемой пробы стали. В четыре стакана (или колбы) приливают последовательно 0,5; 1,0; 3,0; 5,0 см3 стандартного раствора Б. Пятый стакан служит для проведения контрольного (холостого) опыта.
Во все стаканы добавляют по 20 см3 соляной кислоты и 5 см3 азотной кислоты. Далее поступают, как указано в п. 2.3.1 с учетом способа отделения висмута от основных компонентов и в п. 2.3.2.
3. НЕПЛАМЕННЫЙ АТОМНО-АБСОРБЦИОННЫЙ МЕТОД
3.1. Сущность метода
Метод основан на измерении поглощения излучения свободными атомами висмута при длине волны 223,1 или 306,8 нм, образующимися при введении анализируемого раствора в графитовую кювету.
Висмут предварительно отделяют от основных компонентов стали осаждением в виде сульфида тиоацетамидом в аммиачном растворе в присутствии коллектора сульфида меди и винной кислоты в качестве комплексообразующего вещества или методом ионообменной хроматографии.
3.2. Аппаратура, реактивы и растворы
Атомно-абсорбционный спектрофотометр с графитовой кюветой.
Лампа для определения висмута.
Микропипетка вместимостью 20 мкдм3.
Аргон высокой чистоты по ГОСТ 10157 или смесь аргона с 5 % водорода.
Стандартный раствор В висмута: 10 см3 раствора Б, приготовленного по п. 2.2, помещают в мерную колбу вместимостью 100 см3, добавляют 10 см3 азотной кислоты, доливают до метки водой и перемешивают. Раствор готовят непосредственно перед использованием.
1 см3 стандартного раствора В содержит 0,000001 г висмута.
Остальные реактивы, растворы и аппаратура по п. 2.2.
3.3. Проведение анализа
3.3.1. Приготовление испытуемого раствора
Навеску стали массой 0,1 - 1 г согласно табл. 1 помещают в стакан (или колбу) вместимостью 250 - 300 см3, приливают 15 - 20 см3 соляной кислоты, 5 см3 азотной кислоты, накрывают стакан (или колбу) часовым стеклом и растворяют навеску при нагревании.
Таблица 1
|
Масса навески, г |
Объем анализируемого раствора, см3 |
|
|
От 0,0001 до 0,0005 включ. |
1 |
25 |
|
Св. 0,0005 » 0,001 » |
0,5 |
25 |
|
» 0,001 » 0,0025 » |
0,2 |
25 |
|
» 0,0025 » 0,01 » |
0,1 |
50 |
Далее поступают, как указано в п. 2.3.1, отделяя висмут от основных компонентов в виде сульфида тиоацетамидом или методом ионообменной хроматографии.
3.3.2. Спектрометрическая процедура анализа
Испытуемый раствор, приготовленный по п. 2.3.1.1 или 2.3.1.2, выпаривают до влажных солей. Соли растворяют в 5 см3 азотной кислоты и снова выпаривают до влажных солей. Затем соли растворяют при нагревании в 10 см3 азотной кислоты (1:1), накрывая стакан часовым стеклом, и охлаждают. Раствор переносят в мерную колбу (см. табл. 1), доливают водой до метки и перемешивают. Отбирают микропипеткой аликвотную часть раствора, равную 20 мкдм3, вводят в графитовую кювету и фиксируют величину поглощения излучения свободными атомами висмута при длине волны 223,1 или 306,8 нм; для измерения отбирают не менее трех аликвотных частей раствора.
Массу висмута находят по градуировочному графику с учетом поправки контрольного опыта.
3.3.3. Подготовка прибора к измерению
Включение прибора, настройку спектрофотометра на резонансное излучение, регулировку блока управления, блока атомизации проводят согласно инструкции, прилагаемой к прибору.
Условия определения висмута:
Аналитическая линия (λ) - 223,1 или 306,8 нм.
Спектральная ширина щели - 0,2 нм.
Время высушивания при 100 °C - 10 с.
Время разложения при 560 °C - 15 с.
Время атоматизации при 1930 °C - 10 с.
Режим инертного газа «газ-стоп».
3.3.4. Построение градуировочного графика
В пять стаканов (или колб) вместимостью 250 - 300 см3 помещают навески карбонильного железа в количестве, соответствующем массе навески стали (см. табл. 1). В четыре стакана (или колбы) приливают последовательно 1,0; 2,0; 4,0; 5,0 см3 стандартного раствора В висмута. Пятый стакан (или колба) служит для проведения контрольного опыта.
Во все стаканы (или колбы) добавляют по 20 см3 соляной кислоты и 5 см3 азотной кислоты.
Далее поступают, как указано в п. 2.3.1 с учетом способа отделения висмута от основных компонентов и в пп. 3.3.2, 3.3.3.
Из значения оптической плотности анализируемых растворов вычитают значение оптической плотности контрольного опыта. По найденным величинам оптической плотности и соответствующим им массам висмута строят градуировочный график.
4. ИНВЕРСИОННО-ВОЛЬТАМПЕРОМЕТРИЧЕСКИЙ МЕТОД
4.1. Сущность метода
Метод основан на предварительном концентрировании висмута на стационарном ртутном капельном или ртутно-графитовом электроде при потенциале минус 0,5 В в соляной кислоте концентрацией 1 моль/дм3 с последующей регистрацией тока анодного растворения висмута при потенциале минус 0,15 В по отношению к хлорсеребряному электроду.
Висмут отделяют от основных компонентов стали осаждением в виде сульфида тиоацетамидом в аммиачном растворе в присутствии коллектора сульфида меди и винной кислоты в качестве комплексообразующего вещества или методом ионообменной хроматографии.
4.2. Аппаратура, реактивы и растворы
Полярограф переменного тока, осциллографический или постоянного тока.
Ячейка с хлорсеребряным электродом сравнения, стационарным ртутным капельным электродом любой конструкции или твердый дисковый электрод (S ≈ 4 мм2) из графитсодержащего материала любого способа изготовления, обеспечивающие требуемую по НТД воспроизводимость аналитического сигнала.
Потенциостат любой модели, работающий в режиме заданного потенциала.
Ртуть марки Р0 по ГОСТ 4658, не содержащая влаги.
Азот газообразный по ГОСТ 9293 или аргон по ГОСТ 10157.
Фон для полярографирования, содержащий 1 моль/дм3 соляной кислоты. В случае необходимости подвергается дополнительной электрохимической очистке по схеме чертежа от примесей цветных металлов с ртутным катодом в течение 4 - 5 ч при потенциале минус 1,2 В, который поддерживается постоянным с помощью потенциостата.
Калий хлористый по ГОСТ 4234, насыщенный раствор.
Ртуть (II) азотнокислая окисная по ГОСТ 4520, раствор концентрацией 0,001 г/см3 в азотной кислоте (1:15).
Квасцы алюмокалиевые по ГОСТ 4329, раствор концентрацией 10 г/дм3.
Аммоний азотнокислый по ГОСТ 22867.
Стандартные растворы висмута.
Раствор Б: 10 см3 раствора А (по п. 2.2) помещают в мерную колбу вместимостью 100 см3, добавляют 2 см3 соляной кислоты, доливают до метки водой и перемешивают.
1 см3 стандартного раствора Б содержит 0,00001 г висмута.
Раствор В: 5 см3 раствора Б помещают в мерную колбу вместимостью 50 см3, добавляют 2 см3 соляной кислоты, доливают до метки водой и перемешивают.
1 см3 стандартного раствора В содержит 0,000001 г висмута.
Раствор В готовят непосредственно перед использованием.
Остальные реактивы и растворы по п. 2.2.
4.3. Проведение анализа
4.3.1. Приготовление испытуемого раствора
Навеску стали массой 0,5 г помещают в стакан (или колбу) вместимостью 250 - 300 см3, приливают 15 - 20 см3 соляной кислоты, 5 см3 азотной кислоты, накрывают стакан (или колбу) часовым стеклом и растворяют навеску при нагревании.
Далее поступают, как указано в п. 2.3.1, отделяя висмут от основных компонентов в виде сульфида тиоацетамидом (п. 2.3.1.1) или методом ионообменной хроматографии (п. 2.3.1.2).
Испытуемый раствор, полученный по п. 2.3.1.1, выпаривают досуха, соли растворяют в 3 см3 азотной кислоты при нагревании и разбавляют водой приблизительно до 80 см3. К полученному раствору приливают 10 см3 раствора алюмокалиевых квасцов, 0,5 г азотнокислого аммония и аммиак до слабого запаха. Содержимое стакана (или колбы) нагревают в течение 1 - 2 мин, осадок отфильтровывают на фильтр средней плотности (белая лента) и промывают 3 - 4 раза горячим разбавленным (1:200) аммиаком. Фильтрат отбрасывают. Осадок на фильтре растворяют в 10 см3 горячей соляной кислоты (1:1) и промывают фильтр 2 - 3 раза горячей водой, собирая фильтрат и промывные воды в стакан (или колбу), в которых проводилось осаждение.
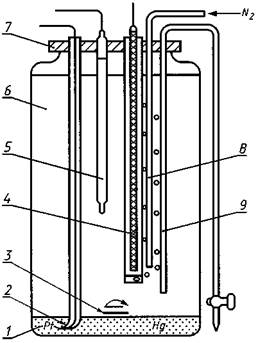
1 - рабочий ртутный электрод; 2 - платиновый
контакт; 3 - магнитная мешалка;
4 - вспомогательный электрод из спектрального угля; 5 -
хлорсеребряный электрод сравнения;
6 - полиэтиленовый сосуд; 7 - крышка; 8 - стеклянная
трубка для подвода азота;
9 - сифон для слива электролита
4.3.2. Инверсионно-вольтамперометрическая процедура анализа
Испытуемый раствор, полученный по п. 4.3.1 после отделения висмута от основных компонентов, выпаривают досуха. Соли растворяют в 5 см3 соляной кислоты и снова выпаривают досуха. Затем соли растворяют при нагревании в 4 см3 соляной кислоты, накрывая стакан часовым стеклом, и охлаждают. Раствор разбавляют водой и переносят в мерную колбу вместимостью 50 см3, доливают водой до метки и перемешивают.
При работе со стационарным ртутным капельным электродом в полярографическую ячейку заливают 20 - 25 см3 фонового электролита, через который предварительно в течение 5 мин продувают азот или аргон, добавляют в соответствии с табл. 2 аликвотную часть испытуемого раствора, 0,01 - 0,02 г аскорбиновой кислоты и перемешивают.
Таблица 2
|
Объем аликвотной части раствора, см3 |
Масса навески стали, соответствующая аликвотной части раствора, г |
|
|
От 0,0001 до 0,0005 включ. |
5 |
0,05 |
|
Св. 0,0005 » 0,001 » |
2 |
0,02 |
|
» 0,001 » 0,005 » |
1 |
0,01 |
Устанавливают на полярографе потенциал минус 0,5 В и проводят концентрирование висмута на стационарном ртутном капельном электроде в непрерывно перемешиваемом растворе в течение 2 - 3 мин. По окончании времени накопления прекращается перемешивание, и раствор успокаивается в течение 15 - 20 с, после чего снимается анодная поляризационная кривая при линейно изменяющемся потенциале электрода от минус 0,5 до минус 0,05 В, регистрируя пик висмута при потенциале минус 0,15 В. Для каждого измерения получают новую каплю ртути.
При работе с твердыми электродами в режиме ртутно-графитового в полярографическую ячейку заливают 20 - 25 см3 фонового электролита, через который предварительно в течение 5 мин продувают азот или аргон, добавляют 3 - 4 капли раствора азотнокислой ртути (II) (150 - 200 мкг), добавляют в соответствии с табл. 3 аликвотную часть испытуемого раствора, 0,01 - 0,02 г аскорбиновой кислоты и перемешивают.
Устанавливают на полярографе потенциал минус 0,5 В и проводят концентрирование висмута на ртутно-графитовом электроде в непрерывно перемешиваемом растворе в течение 1 - 2 мин. По окончании времени накопления прекращается перемешивание, раствор успокаивается в течение 15 - 20 с, после чего снимается анодная поляризационная кривая при линейно изменяющемся потенциале электрода от минус 0,5 до плюс 0,2 В. При фиксированном значении потенциала плюс 0,2 В электрод очищается электрохимически в перемешиваемом растворе в течение 30 с после каждой регистрации поляризационной кривой. Регистрацию кривых проводят три раза, из них первое измерение в расчетах не учитывается. Максимальный ток ионизации висмута (пик висмута) регистрируется при потенциале минус 0,15 В.
Таблица 3
|
Объем аликвотной части раствора, см3 |
Масса навески стали, соответствующая аликвотной части раствора, г |
|
|
От 0,0001 до 0,0005 включ. |
2 |
0,02 |
|
Св. 0,0005 » 0,002 » |
1 |
0,01 |
|
» 0,002 » 0,005 » |
0,5 |
0,005 |
Чувствительность прибора при регистрации вольтамперограмм в обоих случаях выбирают так, чтобы высота регистрируемого пика была не менее 10 мм.
4.3.3. При работе со стационарным ртутным капельным электродом содержание висмута находят по градуировочному графику с учетом контрольного опыта.
Для построения градуировочного графика в пять стаканов (или колб) вместимостью 250 - 300 см3 помещают по 0,5 г карбонильного железа и приливают по 20 см3 соляной кислоты и 5 см3 азотной кислоты. В четыре стакана (или колбы) приливают стандартный раствор В висмута в возрастающих количествах с таким расчетом, чтобы масса висмута в испытуемой пробе стали была приблизительно в середине графика (см. табл. 3). Пятый стакан (или колба) служит для проведения контрольного опыта.
Далее поступают, как указано в пп. 2.3.1 и 4.3.1 с учетом выбранного способа отделения висмута от основных компонентов и п. 4.3.2.
Из значений высоты пика анализируемых растворов вычитают значение высоты пика контрольного опыта. По найденным величинам высоты и соответствующим им массам висмута строят градуировочный график.
При работе с ртутно-графитовым электродом содержание висмута находят методом стандартных добавок.
Аликвотную часть стандартного раствора В висмута добавляют в испытуемый раствор в полярографической ячейке, перемешивают, далее проводят инверсионно-вольтамперометрические измерения (по п. 4.3.2) как при определении висмута в испытуемом растворе.
Величину стандартной добавки выбирают так, чтобы высота пика висмута после введения добавки увеличилась в 1,5 - 2 раза.
5. ПОЛЯРОГРАФИЧЕСКИЙ МЕТОД
5.1. Сущность метода
Метод основан на способности висмута восстанавливаться на ртутном капающем электроде в соляной кислоте концентрацией 1 моль/дм3 при потенциале минус 0,15 В по отношению к хлорсеребряному электроду.
Висмут отделяют от основных компонентов стали осаждением в виде сульфида тиоацетамидом в аммиачном растворе в присутствии сульфида меди и винной кислоты в качестве комплексообразующего вещества или методом ионообменной хроматографии.
5.2. Аппаратура, реактивы и растворы
Полярограф переменного тока, осциллографический или постоянного тока.
Ячейка с ртутным капельным электродом.
Реактивы и растворы по пп. 2.2 и 4.2.
5.3. Проведение анализа
5.3.1. Приготовление испытуемого раствора
Проводят как указано в п. 4.3.1.
5.3.2. Полярографическая процедура анализа
Испытуемый раствор, полученный (п. 4.3.1) после отделения висмута от основных компонентов, выпаривают досуха. Соли растворяют в 5 см3 соляной кислоты и снова выпаривают досуха. Затем соли растворяют при нагревании в 4 см3 соляной кислоты, накрывая стакан часовым стеклом, и охлаждают. Раствор разбавляют водой и переносят в мерную колбу вместимостью 50 см3, доливают водой до метки и перемешивают.
Раствор после продувания инертным газом заливают в ячейку и полярографируют, регистрируя максимальный ток восстановления висмута в пределах приложенного напряжения от 0,05 до 0,3 В относительно хлорсеребряного электрода или ртутного дна.
Чувствительность прибора при регистрации вольтамперограмм выбирают так, чтобы высота пика была не менее 10 мм.
5.3.3. Построение градуировочного графика
В пять стаканов (или колб) вместимостью 250 - 300 см3 помещают по 0,5 г карбонильного железа и приливают по 20 см3 соляной кислоты и 5 см3 азотной кислоты. В четыре стакана (или колбы) приливают стандартный раствор Б висмута в возрастающих количествах с таким расчетом, чтобы масса висмута в испытуемой пробе стали была приблизительно в середине графика. Пятый стакан (или колба) служит для проведения контрольного опыта.
Далее поступают, как указано в пп. 2.3.1 и 4.3.1, с учетом выбранного способа отделения висмута от основных компонентов и в п. 5.3.2.
Из значений высоты пика анализируемых растворов вычитают значение высоты пика контрольного опыта. По найденным величинам высоты и соответствующим им массам висмута строят градуировочный график.
6. ОБРАБОТКА РЕЗУЛЬТАТОВ
6.1. Массовую долю висмута (X) в процентах вычисляют по формулам:
- при расчете по градуировочному графику
![]()
где m - масса висмута, найденная по градуировочному графику, г;
т1 - масса навески стали, г;
- при расчете методом добавок
![]()
где h - высота пика висмута при полярографировании испытуемого раствора, мм;
h1 - высота пика висмута при полярографировании контрольного опыта, мм;
h2 - высота пика висмута после введения в ячейку стандартной добавки, мм;
V - объем стандартной добавки, см3;
с - концентрация стандартного раствора, г/см3;
т - масса навески стали, соответствующая аликвотной части раствора, г.
6.2. Нормы точности и нормативы контроля точности определения массовой доли висмута приведены в табл. 4.
Таблица 4
|
Нормы точности и нормативы контроля точности, % |
|||||
|
D |
dк |
d2 |
d3 |
δ |
|
|
От 0,0001 до 0,0002 включ. |
0,00008 |
0,00010 |
0,00008 |
0,00010 |
0,00005 |
|
Св. 0,0002 » 0,0005 » |
0,00016 |
0,00020 |
0,00017 |
0,00020 |
0,00010 |
|
» 0,0005 » 0,001 » |
0,0004 |
0,0005 |
0,0004 |
0,0005 |
0,0003 |
|
» 0,001 » 0,002 » |
0,0008 |
0,0010 |
0,0008 |
0,0010 |
0,0005 |
|
» 0,002 » 0,005 » |
0,0016 |
0,0020 |
0,0017 |
0,0020 |
0,0010 |
|
» 0,005 » 0,01 » |
0,002 |
0,003 |
0,003 |
0,003 |
0,002 |
ИНФОРМАЦИОННЫЕ ДАННЫЕ
1. РАЗРАБОТАН И ВНЕСЕН Министерством металлургии СССР
2. УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Постановлением Комитета стандартизации и метрологии СССР от 21.10.91 № 1631
3. ВВЕДЕН ВПЕРВЫЕ
4. ССЫЛОЧНЫЕ НОРМАТИВНО-ТЕХНИЧЕСКИЕ ДОКУМЕНТЫ
|
Обозначение НТД, на который дана ссылка |
Номер пункта |
Обозначение НТД, на который дана ссылка |
Номер пункта |
|
Разд. 1 |
|||
|
ТУ 48-6-11-90 |
5. ПЕРЕИЗДАНИЕ. Май 2004 г.