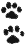 | Информационная система | |

ГОСУДАРСТВЕННЫЙ СТАНДАРТ СОЮЗА ССР
СПЛАВЫ КРЕМНИЕВЫЕ РЕЗИСТИВНЫЕ
ТЕХНИЧЕСКИЕ УСЛОВИЯ
ГОСТ 22025-76
ГОСУДАРСТВЕННЫЙ КОМИТЕТ СССР ПО СТАНДАРТАМ
Москва
ГОСУДАРСТВЕННЫЙ СТАНДАРТ СОЮЗА ССР
|
Технические условия Resistive silicon allons. Specifications |
ГОСТ Взамен |
* Переиздание август 1983 г. с Изменением № 1, утвержденным в марте 1983 г Постановлением № 5110 от 27.12.82 (ИУС № 4-83).
Постановлением Государственного комитета стандартов Совета Министров СССР от 06.03 1976 г. № 1900 срок введения установлен
с 01.01.78
Проверен в 1982 г. Постановлением Госстандарта от 27.12.82 № 5109 срок действия продлен
до 01.01.88
Несоблюдение стандарта преследуется по закону
Настоящий стандарт распространяется на резистивные кремниевые сплавы, предназначенные для изготовления методом вакуумно-термического нанесения термостабильных, тонкопленочных резистивных элементов и различных вспомогательных слоев изделий электронной техники.
1. МАРКИ
1.1. Резистивные кремниевые сплавы выпускают следующих марок: РС-5406, РС-5402, РС-4800, РС-4400, РС-3710, РС-3001, РС-1714, РС-1004.
1.2. Химический состав резистивных кремниевых сплавов должен соответствовать указанному в табл. 1.
|
Химический состав, % по массе |
|||||
|
Основные компоненты |
|||||
|
Хром |
Никель |
Железо |
Вольфрам |
Кобальт |
|
|
РС-5406 |
52,5 - 55,5 |
- |
- |
- |
4,5 - 7,5 |
|
PC-5402 |
52,5 - 55,5 |
- |
1,0 - 3,0 |
- |
- |
|
PC-4800 |
47,0 - 49,0 |
- |
- |
- |
- |
|
PC-4400 |
42,5 - 45,5 |
- |
- |
- |
- |
|
РС-3710 |
36,5 - 39,5 |
8,0 - 11,0 |
- |
- |
- |
|
РС-3001 |
28,0 - 32,0 |
- |
0,7 - 1,8 |
- |
- |
|
PC-1714 |
16,5 - 18,5 |
- |
13,0 - 15,0 |
23,0 - 27,0 |
- |
|
PC-1004 |
- |
9,0 - 42,0 |
3,0 - 6,0 |
- |
- |
Продолжение
|
Химический состав, % по массе |
|||||||
|
Основные компоненты Кремний |
Примеси, не более |
||||||
|
Азот |
Водород |
Кислород |
Углерод |
Алюминий |
Медь |
||
|
РС-5406 |
Остальное |
0,02 |
0,003 |
0,30 |
0,06 |
- |
- |
|
РС-5402 |
То же |
0,02 |
0,003 |
0,30 |
0,06 |
- |
- |
|
РС-4800 |
» |
0,02 |
0,003 |
0,30 |
0,06 |
- |
- |
|
РС-4400 |
» |
0,02 |
0,003 |
0,30 |
0,06 |
- |
- |
|
РС-3710 |
» |
0,02 |
0,003 |
0,30 |
0,06 |
- |
- |
|
РС-3001 |
» |
0,02 |
0,003 |
0,30 |
0,06 |
- |
- |
|
РС-1714 |
» |
- |
0,005 |
0,60 |
0,05 |
0,20 |
0,01 |
|
РС-1004 |
» |
0,02 |
0,003 |
0,30 |
0,0,6 |
- |
- |
1.3. Условное обозначение резистивных кремниевых сплавов при заказе и в технической документации должно состоять из слова сплав, марки (сокращенного обозначения резистивного сплава и четырех цифр: две первые цифры - номинальное содержание одного легирующего компонента, а две последующие - номинальное содержание другого легирующего компонента) номера фракции и обозначения настоящего стандарта.
Пример записи резистивных кремниевых сплавов:
Сплав РС-4800 Фр. 1 ГОСТ 22025-76
1.1 - 1.3. (Измененная редакция, Изм. № 1).
2. ТЕХНИЧЕСКИЕ ТРЕБОВАНИЯ
2.1. Резистивные кремниевые сплавы должны изготовляться в соответствии с требованиями настоящего стандарта по технической документации, утвержденной в установленном порядке.
2.2. Резистивные кремниевые сплавы выпускают в виде порошков темно-серого или бурого цвета с размерами частиц:
не более 0,040 мм - для фракции 1;
от 0,040 до 0,071 мм - для фракции 2;
от 0,094 до 0,140 мм (только для марки РС-3710) - для фракции 3.
(Измененная редакция, Изм. № 1).
2.3. Количество порошка резистивных кремниевых сплавов (гранулометрический состав) с размерами частиц, выходящими за пределы, указанные в п. 2.2, не должно превышать 5 % от массы навески, взятой от средней пробы.
2.4. Порошки резистивных кремниевых сплавов не должны содержать посторонних включений.
2.5. Время истечения порошка резистивного кремниевого сплава, заключенного между верхней и нижней отметками воронки (черт. 1), не должно превышать:
60 с - для фракции 1;
35 с - для фракции 2;
30 с - для фракции 3.
Время истечения порошка марки РС-4800 1-й фракции не должно превышать 50 с.
Время истечения порошка сплава марки РС-1714 не устанавливают.
(Измененная редакция, Изм. № 1).
2.6. Порошок резистивного кремниевого сплава марки РС-1714 должен смачиваться этиловым ректификованным спиртом по ГОСТ 18300-72, образуя однородную суспензию.
2.7. Данные о физических и электрических параметрах резистивных кремниевых сплавов в объемном состоянии и в виде пленки приведены в справочном приложении 1.
2.8. Область применения и методы нанесения резистивных кремниевых сплавов даны в рекомендуемом приложении 2.
3. ПРАВИЛА ПРИЕМКИ
3.1. Резистивные кремниевые сплавы предъявляют к приемке партиями. В партию входит сплав одной марки, полученный от одного или нескольких технологических циклов при одном технологическом режиме.
Масса партии не должна превышать 10 кг.
3.2. Каждую партию резистивных кремниевых сплавов подвергают приемо-сдаточным испытаниям для проверки соответствия химического состава, сыпучести и смачиваемости требованиям настоящего стандарта.
Содержание кремния определяют только в случае изменений в технологическом процессе их производства. В остальных случаях содержание кремния определяют как разность между 100 % химического состава сплава и суммой содержания остальных определяемых компонентов сплава, выраженную в процентах.
Размеры частиц определяют выборочно на одной из пяти партий.
Проверку сплавов на содержание примесей проводят по требованию предприятия-потребителя.
3.3. Все испытания резистивных кремниевых сплавов проводят на средней пробе от каждой партии.
3.4. При получении неудовлетворительных результатов хотя бы по одному из показателей проводят повторную проверку новой пробы, взятой от той же партии сплава. Результаты повторных испытаний являются окончательными и распространяются на всю партию.
Воронка для определения времени истечения порошков резистивных кремниевых сплавов
4. МЕТОДЫ ИСПЫТАНИЙ
4.1. Метод отбора проб
4.1.1. Среднюю пробу отбирают в боксе, исключая загрязнения и потери порошка.
4.1.2. Всю партию порошка высыпают в квадратный противень из нержавеющей стали, который предварительно выстилают полиэтиленовой пленкой по ГОСТ 10354-82, вымытой дистиллированной водой по ГОСТ 6709-72 и протертой этиловым спиртом по ГОСТ 18300-72.
Порошок перемешивают методом «кольцо на конус» стальным хромированным шпателем и распределяют по поверхности противня ровным слоем, после чего делят на 25 равных квадратов и отбирают порошок по всей толщине слоя из 13 квадратов в шахматном порядке примерно одинаковыми порциями.
Масса отобранного порошка должна быть не менее 110 г.
Из отобранного порошка приготовляют следующие пробы: 50 г для проверки гранулометрического состава, 20 г для определения времени истечения порошка, две примерно равные по массе пробы из оставшейся части порошка - одну для химического анализа, другую для хранения в течение гарантийного срока на случай проведения арбитражных испытаний при возникновении разногласий при оценке качества порошка.
Все пробы засыпают в двойные пакеты из полиэтиленовой пленки, предварительно протертые этиловым спиртом. Последние две пробы заваривают герметичным швом.
(Измененная редакция, Изм. № 1, 2).
4.1.3. Между внутренними и наружными пакетами вкладывают этикетку, на которой указывают:
номер цеха-изготовителя;
наименование сплава;
номер партии и пробы;
размер частиц порошка;
массу пробы;
дату отбора пробы (месяц, год).
4.2. Определение гранулометрического состава
Испытания проводят на контрольных ситах с металлическими проволочными сетками из нержавеющей стали 0040К для фракции 1, 0040К и 0071К для фракции 2 (по нормативно-технической документации, утвержденной в установленном порядке) и 009К (по ГОСТ 6613-86) и 014К для фракции 3, установленных на вибровстряхиватель (частота вибрации 1500 - 3000 колебаний в минуту, амплитуда вибрации 0,1 - 0,2 см).
Навеску порошка массой (50 ± 0,1) г просеивают в течение 20 мин через сито с сеткой 0040К для сплавов 1-й фракции или через набор сит с сетками 0040К и 0071 К, 009К и 014К для сплавов фракции 2 и 3 соответственно.
Количество порошка, оставшегося на сите с сеткой 0040К для сплава фракции 1, или суммарное количество порошка, оставшегося на сите с сеткой 0071К и прошедшего через сито с сеткой 0040К для сплавов фракции 2, и оставшегося на сите с сеткой 014К и прошедшего через сито с сеткой 0091К для сплавов фракции 3, не должно превышать 2,5 г.
Погрешность взвешивания должна быть не более 0,1 г.
(Измененная редакция, Изм. № 1, 2).
4.3. Определение содержания посторонних включений
Содержание посторонних включений кремниевых сплавов проверяют визуально невооруженным глазом, а также при помощи микроскопа МБС-1 при шестнадцатикратном увеличении.
4.4. Определение времени истечения порошка
4.4.1. Аппаратура, материалы:
шкаф сушильный вакуумный ВШ-0,035;
воронка из молибденового стекла марки С49-2 (см. черт. 1);
штатив лабораторный типа ШЛ;
секундомер СДПпр-1б-2 по ГОСТ 5072-79;
ацетон по ГОСТ 2603-79, ч.;
стакан типа стакан В-1-100ТС по ГОСТ 25336-82;
ткань хлопчатобумажная, арт. 324 по ГОСТ 7138-83.
(Измененная редакция, Изм. № 1, 2).
4.4.2. Подготовка к испытанию
Воронку и стакан предварительно промывают ацетоном и протирают чистой тканью.
Воронку устанавливают в штативе в вертикальном положении по отвесу, под ней помещают стакан. Расстояние от воронки до стакана не должно превышать 30 мм.
4.4.3. Проведение испытания
Порошок из пакета высыпают в воронку выше верхней отметки и засекают по секундомеру моменты прохождения поверхностью порошка верхней и нижней отметок. Замеры времени повторяют не менее пяти раз. За результат измерения принимают среднее арифметическое результатов замеров.
Струя высыпающегося порошка должна быть непрерывной. Не допускается постукивать по воронке в момент проведения испытания.
4.5. Определение содержания хрома объемным персульфатносеребряным методом в сплавах марок РС-5406, РС-5402, РС-4800, РС-4400, РС-3710 и РС-3001
4.5.1. Сущность метода
Метод основан на окислении хрома надсернокислым аммонием в сернокислой среде до бихромат-иона в присутствии азотнокислого серебра. Бихромат-ион титруют солью Мора с фенилантраниловой кислотой в качестве индикатора.
Относительная погрешность результата измерения должна быть не более 0,5 %.
4.5.2. Реактивы, материалы:
чашка ПЛ 118-4 по ГОСТ 6563-75;
крышки типа «часовое стекло» из фторопласта-4 по ГОСТ 10007-80;
аммоний надсернокислый по ГОСТ 20478-75, х. ч.;
вода дистиллированная по ГОСТ 6709-72;
калий двухромовокислый, стандарт-титр, 0,1 н. раствор;
кислота азотная по ГОСТ 4461-77, х. ч.;
кислота серная по ГОСТ 4204-77, х. ч.;
кислота фтористоводородная по ГОСТ 10484-78, х. ч.;
марганец (II) сернокислый 5-водный по ГОСТ 435-77, ч. д.а., 0,4 %-ный раствор;
натрий углекислый 10-водный по ГОСТ 84-76, х. ч.;
натрий хлористый по ГОСТ 4233-77. х. ч., 5 %-ный раствор;
серебро азотнокислое по ГОСТ 1277-75, х. ч., 1 %-ный раствор;
соль закиси железа и аммония двойная сернокислая (соль Мора) по ГОСТ 4208-72, х. ч., 0,1 н. раствор; готовят следующим образом: 40 г соли Мора растворяют в 200 мл воды, подкисленной 30 мл серной кислоты, и разбавляют водой до объема 1000 мл. Нормальность этого раствора устанавливают по 0,1 н. раствору двухромовокислого калия в день титрования образца;
кислота N-фенилантраниловая, ч. д. а., 0,2 %-ный раствор; готовят следующим образом: 0,2 г N-фенилантраниловой кислоты растворяют при умеренном нагревании в 30 - 40 мл воды, содержащей 0,6 г углекислого натрия, разбавляют водой до объема 100 мл. Хранят в темной склянке или темном месте.
(Измененная редакция, Изм. № 2).
4.5 - 4.5.1. (Измененная редакция, Изм. № 1).
4.5.3. Проведение испытания
0,1 г анализируемого сплава взвешивают с погрешностью не более 0,0001 г и помещают в платиновую чашку. Добавляют 2 мл дистиллированной воды, 5 мл фтористоводородной кислоты, накрывают чашку фторопластовой крышкой и, периодически приподнимая крышку, по каплям добавляют азотную кислоту до растворения сплава.
По окончании растворения крышку обмывают дистиллированной водой, осторожно добавляют 10 мл серной кислоты и выпаривают раствор до обильного выделения паров серного ангидрида. Содержимое чашки охлаждают, переносят в коническую колбу вместимостью 250 мл и разбавляют дистиллированной водой до объема 100 мл.
Добавляют в колбу 1 мл раствора сернокислого марганца, 5 мл раствора азотнокислого серебра, 5 г надсернокислого аммония и нагревают раствор до кипения. Нагревание продолжают до прекращения выделения мелких пузырьков кислорода, т.е. до полного разложения надсернокислого аммония. Окисление хрома считают законченным, когда появится устойчивая красно-фиолетовая окраска перманганат-йона. Если эта окраска отсутствует, осторожно прибавляют еще несколько порций (массой около 0,5 г) надсернокислого аммония и нагревают до появления окраски и прекращения выделения пузырьков кислорода. Добавляют около 10 мл раствора хлористого натрия до исчезновения окраски перманганат-йона и оставляют на теплой плите до просветления желтого раствора. Раствор охлаждают, добавляют пять-шесть капель N-фенилантраниловой кислоты и титруют раствором соли Мора до перехода грязновато-коричневой окраски через красно-фиолетовую в зеленую.
4.5.4. Обработка результатов
Содержание хрома (X) в процентах вычисляют по формуле
![]() ,
,
где Т - титр точно 0,1 н. раствора соли Мора по хрому, 0,001733 г/мл;
V - объем раствора соли Мора, израсходованный на титрование, мл;
m - масса навески анализируемого сплава, г.
Для каждой пробы проводят не менее двух параллельных определений.
4.6. Определение содержания никеля весовым методом
4.6.1. Сущность метода
Никель осаждают из аммиачного раствора в присутствии лимонной кислоты диметилглиоксимом, фильтруют через стеклянный тигель с фильтрующим дном, высушивают и взвешивают в виде диметилглиоксимата никеля. Относительная погрешность определения не должна быть более 1 %.
4.6.2. Аппаратура, реактивы и растворы:
тигель ТФ-10-ПОР-10 ХС по ГОСТ 25336-82;
аммиак водный по ГОСТ 3760-79, х. ч.;
вода дистиллированная по ГОСТ 6709-72;
диметилглиоксим по ГОСТ 5828-77, ч. д. а., 1 %-ный спиртовой раствор;
кислота азотная по ГОСТ 4461-77, х. ч.;
кислота лимонная по ГОСТ 3652-69, х. ч., 25 %-ный раствор;
кислота серная по ГОСТ 4204-77, х. ч.;
кислота фтористоводородная по ГОСТ 10484-78, х. ч.;
спирт этиловый ректификованный технический, высший сорт по ГОСТ 18300-72;
чашки ПЛ 118-4 по ГОСТ 6563-75;
крышки типа «часовое стекло» из фторопласта-4 по ГОСТ 10007-80.
(Измененная редакция, Изм. № 1, 2).
4.6.3. Проведение испытания
Навеску сплава массой около 0,25 г взвешивают с погрешностью не более 0,0001 г и помещают в платиновую чашку. Прибавляют 0,5 мл дистиллированной воды, 5 мл фтористоводородной кислоты, накрывают чашку фторопластовой крышкой и периодически, по каплям добавляя азотную кислоту, растворяют сплав. Обмывают крышку водой, прибавляют 5 мл серной кислоты, осторожно перемешивают и выпаривают раствор до выделения густых паров серного ангидрида. Раствор охлаждают, переносят в стакан вместимостью 300 мл, прибавляют 30 мл раствора лимонной кислоты, нагревают до температуры 40 - 45 °С, прибавляют 25 мл раствора диметилглиоксима и аммиак до слабого запаха.
Раствор с осадком нагревают до температуры 60 - 70 °С, выдерживают при этой температуре 30 мин и фильтруют через предварительно высушенный и взвешенный тигель с фильтрующим дном № 3. Осадок промывают 10 - 12 раз горячей водой и просушивают при температуре 110 - 115 °С до постоянной массы.
4.6.4. Обработка результатов
Содержание никеля (Х1) в процентах вычисляют формуле
![]()
где m1 - масса осадка диметилглиоксимата никеля, г;
m - масса навески образца, г;
0,2032 - коэффициент пересчета диметилглиоксимата никеля на никель.
Для каждой пробы проводят не менее двух параллельных определений.
4.7. Определение содержания железа фотометрическим методом в сплаве марок РС-3001 и РС-5402
4.7.1. Сущность метода
Метод распространяется на резистивные кремниевые сплавы, содержащие железа до 3 %.
Железо определяют фотометрическим методом с сульфосалициловой кислотой после предварительного окисления хрома перекисью водорода в аммиачной среде и отделении гидроокиси железа. Относительная погрешность определения не должна быть более 2,5 %.
4.7.2. Аппаратура, реактивы:
спектрофотометр типа СФ-16 или колориметр фотоэлектрический типа ФЭК-60; допускается применять спектрофотометры и фотоэлектроколориметры других аналогичных марок;
аммиак водный по ГОСТ 3760-79, х. ч.;
аммоний хлористый по ГОСТ 3773-72, х. ч., 20 %-ный раствор;
вода дистиллированная по ГОСТ 6709-72;
водорода перекись по ГОСТ 10929-76;
железо карбонильное радиотехническое по ГОСТ 13610-79;
кислота азотная по ГОСТ 4461-77, х. ч.;
кислота серная по ГОСТ 4204-77, х. ч.;
кислота соляная по ГОСТ 3118-77, х. ч., раствор разбавленный 1:1;
кислота сульфосалициловая по ГОСТ 4478-78, ч., 20 %-ный раствор;
кислота фтористоводородная по ГОСТ 10484-78, х. ч.;
метиловый красный ч.д.а., 0,2 %-ный водно-спиртовой раствор;
стандартный раствор железа 10 мкг/мл, готовят из запасного раствора, содержащего 1 мг/мл, разбавлением в 100 раз;
запасной раствор; готовят растворением 1 г железа, содержащего 99,9 % железа, при нагревании в соляной кислоте. Полученный раствор переносят в мерную колбу вместимостью 1000 мл и доливают водой до полного объема. Полученный раствор содержит 1 мг/мл железа;
чашки ПЛ 118-4 по ГОСТ 6563-75;
фильтр обеззоленный «Красная лента»;
крышка типа «часовое стекло» из фторопласта-4 по ГОСТ 10007-80.
(Измененная редакция, Изм. № 1, 2).
4.7.3. Проведение испытания
Навеску сплава массой около 0,1 г взвешивают с погрешностью не более 0,0001 г и помещают в платиновую чашку, добавляют 2 мл дистиллированной воды, 5 мл фтористоводородной кислоты, покрывают фторопластовой крышкой и осторожно, по каплям, добавляют азотную кислоту до полного растворения сплава. Крышку снимают, обмывают дистиллированной водой, приливают 7 - 10 мл концентрированной серной кислоты и выпаривают до появления паров серного ангидрида. Чашку охлаждают, обмывают стенки водой и снова выпаривают до появления паров серного ангидрида. Содержимое чашки смывают водой в стакан вместимостью 300 мл, добавляют 5 мл соляной кислоты, 15 - 20 мл перекиси водорода, две-три капли метилового красного и 20 - 30 мл раствора аммиака порциями при тщательном перемешивании, нагревают до коагуляции осадка гидроокиси железа и фильтруют через фильтр «красная лента». Осадок промывают пять-шесть раз горячим раствором хлористого аммония, содержащим одну - две капли аммиака на 1000 мл раствора. Промытый осадок растворяют в 10 мл соляной кислоты, переносят раствор в мерную колбу вместимостью 100 мл и доводят до полного объема водой.
Аликвотную часть раствора объемом 1 - 10 мл, содержащую 15 - 40 мкг железа, отбирают в мерную колбу вместимостью 50 мл, добавляют 5 мл сульфосалициловой кислоты, несколько капель раствора аммиака до появления желтой окраски раствора и, в избыток, 5 мл аммиака, доводят водой до полного объема и перемешивают.
Замеряют оптическую плотность раствора на спектрофотометре при длине волны 420 - 450 нм или на колориметре со светофильтром № 3 в кювете с толщиной поглощающего свет слоя 30 мм относительно раствора, не содержащего железо (нулевого раствора). Одновременно с проведением испытания проводят холостой опыт.
Содержание железа в 50 мл раствора определяют по калибровочному графику.
4.7.4. Построение калибровочного графика
Калибровочную график строят в координатах: по оси ординат - оптическая плотность раствора, скорректированная на холостой опыт, по оси абсцисс - количество железа в микрограммах в 50 мл раствора.
В мерные колбы вместимостью по 50 мл каждая помещают проведенные через все стадии анализа 0,0; 0,5; 1,0; 1,5; 2,0; 3,0; 4,0; 5,0 мл стандартного раствора, содержащего соответственно 0; 5; 10; 15; 20; 30; 40; 50 мкг железа.
Добавляют 5 мл раствора сульфосалициловой кислоты, несколько капель раствора аммиака до появления желтой окраски раствора и, в избыток, 5 мл аммиака. Доливают водой до полного объема и перемешивают.
Оптическую плотность раствора измеряют относительно нулевого раствора на спектрофотометре при длине волны 430 нм или фотоэлектроколориметре со светофильтром, имеющим максимальное пропускание в области 420 - 450 нм, в кювете с толщиной поглощающего свет слоя 30 мм.
4.7.5. Обработка результатов
Содержание железа (Х2) в процентах вычисляют по формуле
![]()
где С - количество железа, найденное по калибровочному графику, мкг/50 мл;
m - масса навески образца, соответствующая аликвоте, г.
4.8. Определение содержания железа в сплаве марки РС-1004 фотометрическим методом
4.8.1. Сущность метода
Железо определяют фотометрическим методом с сульфосалициловой кислотой в аммиачной среде без отделения его от никеля. Относительная погрешность определения должна быть не более 2,5 %.
4.8.2. Аппаратура, реактивы и растворы:
спектрофотометр типа СФ-16 или колориметр фотоэлектрический типа ФЭК-60; допускается применять спектрофотометры или фотоэлектроколориметры других аналогичных марок;
аммиак водный по ГОСТ 3760-79, х. ч.;
вода дистиллированная по ГОСТ 6709-72;
железо карбонильное радиотехническое по ГОСТ 13610-79;
кислота азотная по ГОСТ 4461-77, х. ч.;
кислота серная по ГОСТ 4204-77, х.ч.;
кислота сульфосалициловая по ГОСТ 4478-78, ч., 20 %-ный раствор;
кислота фтористоводородная по ГОСТ 10484-78, х. ч.;
никель хлористый по ГОСТ 4038-79, х. ч., раствор, содержащий 0,016 мг/мл;
стандартный раствор железа 10 мкг/мл; готовят из запасного раствора, содержащего 1 мг/мл, разбавлением в 100 раз;
запасной раствор; готовят растворением 1 г железа, содержащего 99,9 % железа, в соляной кислоте при нагревании. Полученный раствор переносят в мерную колбу вместимостью 1000 мл и доводят до полного объема водой. Полученный раствор содержит 1 мг/мл железа.
(Измененная редакция, Изм. № 1).
4.8.3. Проведение испытания
Навеску сплава массой около 0,1 г взвешивают с погрешностью не более 0,0001 г и помещают в платиновую чашку. Добавляют 2 мл дистиллированной воды, 5 мл фтористоводородной кислоты, накрывают крышкой и осторожно по каплям добавляют азотную кислоту до полного растворения сплава. Обмывают крышку дистиллированной водой, приливают 3 - 5 мл серной кислоты и выпаривают до паров серного ангидрида. Чашку охлаждают, обмывают стенки водой и снова выпаривают в мерную колбу вместимостью 100 мл и доводят до полного объема водой.
1 - 10 мл аликвотной части раствора, содержащей 10 - 40 мкг железа, отбирают в мерную колбу вместимостью 50 мл, добавляют 5 мл сульфосалициловой кислоты, несколько капель раствора аммиака до появления желтой окраски и, в избыток, 5 мл аммиака, доводят до полного объема водой и перемешивают.
Оптическую плотность раствора измеряют на спектрофотометре при длине волны 420 - 450 нм или на колориметре со светофильтром № 3 в кювете с толщиной поглощающего свет слоя 30 мм относительно нулевого раствора.
Одновременно с проведением испытания проводят холостой опыт со всеми применяемыми реактивами.
Количество железа определяют по калибровочному графику.
4.8.4. Построение калибровочного графика
Калибровочный график строят в координатах: по оси ординат - оптическая плотность раствора, скорректированная на холостой опыт, по оси абсцисс - количество железа в микрограммах в 50 мл раствора.
В мерные колбы вместимостью по 50 мл каждая помещают проведенные через все стадии анализа 0,0; 0,5; 1,0; 1,5; 2,0; 3,0; 4,0; 5,0 мл стандартного раствора, содержащего соответственно 0,5; 10; 15; 20; 30; 40; 50 мкг железа.
Добавляют 5 мл раствора хлористого никеля, 5 мл сульфосалициловой кислоты, несколько капель раствора аммиака до появления желтой окраски раствора и в избыток 5 мл аммиака. Доливают до полного объема водой и перемешивают.
Оптическую плотность раствора измеряют относительно нулевого раствора на спектрофотометре при длине волны 430 нм или фотоэлектроколориметре со светофильтром, имеющим максимальное пропускание в области 420 - 450 нм, в кювете с толщиной поглощающего свет слоя 30 мм.
4.8.5. Обработка результатов
Содержание железа (Х3) в процентах вычисляют по формуле
![]()
где С - количество железа, найденное по калибровочному графику, мкг/мл;
m - масса навески образца/ соответствующая аликвоте, г.
4.9. Определение содержания хрома, железа и вольфрама в сплаве марки РС-1714 рентгеноспектральным методом
4.9.1. Сущность метода
Метод основан на линейной зависимости интенсивностей флуоресцентного рентгеновского излучения от весовой концентрации анализируемого элемента в образце. На рентгеновском флуоресцентном спектрометре определяют интенсивности флуоресцентного излучения анализируемого и стандартного образцов для аналитических линий хрома, железа и вольфрама. Сравнивая эти интенсивности для каждого из определяемых элементов, рассчитывают их процентное содержание в анализируемом образце.
Коэффициент вариации измерений состава сплава не превышает 10 % для хрома, 8 % для железа, 6 % для вольфрама.
4.9.2. Аппаратура
Для проведения испытания применяют один из указанных ниже приборов;
флуоресцентный рентгеновский квантометр типов ФРК-1Б и КРФ-П;
аппарат рентгеновский типа КРФ-18;
спектрометр рентгеновский флуоресцентный фирмы «Элиот», модель XZ-1030, снабженный запасной рентгеновской трубкой с вольфрамовым анодом производства фирмы «Махлет».
4.9.3. Проведение испытания
Навеску сплава массой не менее 16 г делят на четыре равные части и помещают в четыре кюветы.
В качестве стандартного образца берут тот же сплав, проанализированный методом химического анализа. Химический состав стандартного образца не должен отличаться от анализируемого по каждому из элементов более чем на 15 %. Навеску стандартного образца массой не менее 20 г помещают в две кюветы.
На рентгеновском спектрометре определяют интенсивности флуоресценции двух кювет со стандартным образцом и четырех кювет с анализируемым образцом.
Интенсивности флуоресценции измеряют по аналитическим линиям СrКa, FeKa, WKa.
4.9.4. Обработка результатов
В зависимости от интенсивности флуоресценции по каждой из двух кювет со стандартным образцом и четырех кювет с анализируемым образцом вычисляют содержание определяемого элемента (Х4) в процентах по формуле
![]()
где ![]() - содержание
определяемого элемента в стандартном образце, %;
- содержание
определяемого элемента в стандартном образце, %;
Iэ, Iо - интенсивность флуоресценции от анализируемого и стандартного образцов соответственно, имп.
Для каждого определяемого элемента получают восемь значений концентраций. Среднее этих восьми значений дает искомую величину концентрации определяемого элемента в анализируемом образце.
4.10. Определение содержания хрома объемным персульфатносеребряным методом в сплаве марки РС-1714
4.10.1. Сущность метода
Метод основан на окислении хрома надсернокислым аммонием в сернокислой среде до бихромат-иона в присутствии азотнокислого серебра. Бихромат-ион титруют солью Мора с N-фенилантраниловой кислотой в качестве индикатора.
Относительная погрешность определения должна быть не более 1 %.
(Измененная редакция, Изм. № 1).
4.10.2. Материалы, реактивы:
чашка ПЛ 118-4 по ГОСТ 6563-75;
аммоний надсернокислый по ГОСТ 20478-75, х. ч.;
вода дистиллированная по ГОСТ 6709-72;
калий двухромовокислый, стандарт-титр, 0,1 н. раствор;
кислота азотная по ГОСТ 4461-77, х. ч.;
кислота борная по ГОСТ 9656-75, х. ч., 5 %-ный раствор;
кислота серная по ГОСТ 4204-77, х. ч., разбавленная 1:1;
кислота фтористоводородная по ГОСТ 10484-78, х. ч.;
кислота N-фенилантраниловая, ч. д. а., 0,1 %-ный раствор, готовят по ГОСТ 4919.1-77;
марганец сернокислый по ГОСТ 435-77, ч. д. а., 0,4 %-ный раствор;
натрий хлористый по ГОСТ 4233-77, х. ч., 5 %-ный раствор;
серебро азотнокислое по ГОСТ 1277-75, х. ч., 1 %-ный раствор;
соль закиси железа и аммония двойная сернокислая (соль Мора) по ГОСТ 4208-72, х. ч., 0,05 н. раствор; готовят следующим образом: 20 г соли Мора растворяют в 500 мл воды, содержащей 100 мл серной кислоты, раствор разбавляют водой до объема 1000 мл и фильтруют. Хранят в склянке с притертой пробкой. Нормальность раствора устанавливают в день употребления по 0,1 н. раствору двухромовокислого калия следующим образом: 10 мл раствора двухромовокислого калия помещают в коническую колбу вместимостью 250 мл, приливают 8 мл серной кислоты, 4 мл фосфорной кислоты, воду до объема 150 мл, три капли раствора N-фенилантраниловой кислоты, перемешивают и титруют раствором соли Мора до исчезновения фиолетового окрашивания индикатора.
Коэффициент нормальности раствора соли Мора (К) рассчитывают по формуле
![]()
где N - нормальность раствора двухромовокислого калия;
V1 - объем раствора соли Мора, израсходованного на титрование, мл;
V - объем раствора двухромовокислого калия, взятого на титрование, мл.
(Измененная редакция, Изм. № 1, 2).
4.10.3. Проведение испытания
Навеску сплава массой около 0,1 г взвешивают с погрешностью не более 0,0001 г, помещают в платиновую чашку, смачивают 2 мл воды, добавляют 2 - 3 мл фтористоводородной кислоты, перемешивают и оставляют на 15 - 20 мин при комнатной температуре. Приливают небольшими порциями 1 мл азотной кислоты и продолжают растворение сплава при умеренном нагревании. Раствор выпаривают до объема около 2 мл. Обмывают стенки чашки водой, раствор перемешивают и снова выпаривают до объема 2 мл. Обработку водой повторяют. Раствор в чашке разбавляют водой и переносят в коническую колбу вместимостью 250 мл. Добавляют воду до объема 100 мл, 10 мл раствора борной кислоты, 10 мл серной кислоты, 1 мл раствора сернокислого марганца, 5 мл раствора азотнокислого серебра, 5 г надсернокислого аммония и нагревают раствор почти до кипения. Нагревание продолжают до прекращения выделения мелких пузырьков кислорода, т.е. до полного разложения надсернокислого аммония. Окисление хрома считают законченным тогда, когда появится устойчивая красно-фиолетовая окраска перманганат-йона. Если эта окраска отсутствует, осторожно прибавляют еще несколько порций (массой около 0,5 г) надсернокислого аммония и нагревают до появления красно-фиолетовой окраски и прекращения выделения пузырьков кислорода.
Приливают около 10 мл раствора хлористого натрия до исчезновения окраски перманганат-йона и оставляют на теплой плите до просветления раствора. Раствор охлаждают, добавляют пять-шесть капель раствора N-фенилантраниловой кислоты и титруют раствором соли Мора до перехода буро-коричневой окраски раствора в зеленую.
4.10.4. Обработка результатов
Содержание хрома (Х5) в процентах рассчитывают по формуле
![]()
где Т - титр точно 0,05 н. раствора соли Мора по хрому, 0,000867 г/мл;
К - коэффициент нормальности раствора соли Мора;
V - объем раствора соли Мора, израсходованный на титрование, мл;
m - масса навески сплава, г.
4.11. Определение железа в сплаве марки РС-1714.
4.11.1. Сущность метода
Метод основан на комплексометрическом титровании железа при рН 2 - 3 с сульфосалициловой кислотой в качестве индикатора. Железо предварительно отделяют от вольфрама и хрома осаждением его в виде гидроокиси едким натром в присутствии перекиси водорода.
Относительная погрешность определения должна быть не более 1,5 %.
4.11.2. Материалы, реактивы:
чашки ПЛ 118-4 по ГОСТ 6563-75;
фильтр обеззоленный «красная лента»;
аммиак водный по ГОСТ 3760-79, х. ч.,
25 %-ный раствор; аммоний уксуснокислый по ГОСТ 3117-78, х. ч.;
аммонии хлористый по ГОСТ 3773-72, х. ч.;
буферный раствор рН 9,5 - 10; готовят следующим образом: 54 г хлористого аммония растворяют в 200 мл деионизованной воды, прибавляют 350 мл раствора аммиака и разбавляют водой до объема 1000 мл;
вода деионизованная марки Б;
вода дистиллированная по ГОСТ 6709-72;
водорода перекись по ГОСТ 10929-76;
калий хлористый по ГОСТ 4234-77, х. ч.;
кислота азотная по ГОСТ 4461-77, х. ч., концентрированная и разбавленная 1:1;
кислота серная по ГОСТ 4204-77, х. ч.;
кислота соляная по ГОСТ 3118-77, х. ч.;
кислота сульфосалициловая по ГОСТ 4478-78, х. ч., 10 %-ный раствор;
кислота фтористоводородная по ГОСТ 10484-78, х. ч.;
натрия гидроокись по ГОСТ 4328-77, х. ч., 25 %-ный раствор;
трилон Б (этилендиамин-N,N,N',N'-тетрауксусной кислоты динатриевая соль) по ГОСТ 10652-73, ч. д. а., 0,025 М раствор. Готовят следующим образом: 9,3 г препарата растворяют в деионизованной воде и разбавляют раствор водой до объема 1000 мл. Молярность раствора трилона Б устанавливают по раствору хлористого цинка следующим образом: 20 мл раствора хлористого цинка помещают в коническую колбу вместимостью 250 мл, приливают 15 мл буферного раствора, деионизованную воду до объема 100 мл, 0,05 - 0,1 г индикаторной смеси эриохрома черного и титруют раствором трилона Б до перехода красно-фиолетовой окраски раствора в синюю. Коэффициент полярности раствора трилона Б (К1) рассчитывают по формуле
![]()
где М - молярность раствора хлористого цинка;
V - объем раствора хлористого цинка, взятого для титрования, мл;
V1 - объем раствора трилона Б, израсходованный на титрование, мл;
цинк металлический ЦВ-00 по ГОСТ 3640-79;
цинк хлористый, 0,025 М раствор. Готовят следующим образом: 1,6345 г металлического цинка растворяют в 25 мл соляной кислоты, разбавленной 1:1, раствор разбавляют деионизованной водой в мерной колбе вместимостью 1000 мл;
эриохром черный Т, ч. д. а., индикаторная смесь с хлористым калием 1:100.
(Измененная редакция, Изм. № 1, 2).
4.11.3. Проведение испытания
Навеску сплава массой 0,1 - 0,15 г, взвешенную с погрешностью не более 0,0001 г, помещают в платиновую чашку, смачивают 1 - 2 мл воды, приливают 2 - 3 мл фтористоводородной кислоты, перемешивают и оставляют на 15 - 20 мин при комнатной температуре. Добавляют порциями, перемешивая 5 мл азотной кислоты, и нагревают до полного растворения сплава. Раствор выпаривают до влажного остатка. Приливают 5 мл азотной кислоты и снова выпаривают до влажного остатка. Последнюю операцию повторяют. К остатку приливают 5 мл разбавленной 1:1 азотной кислоты и содержимое чашки переносят в стакан вместимостью 300 - 400 мл, смывая чашку водой. Приставший к стенкам чашки осадок растворяют несколькими каплями аммиака и смывают водой в тот же стакан. Раствор разбавляют водой до объема около 80 мл, приливают 20 - 30 мл пергидроля и раствор едкого натра до выпадения осадка гидроокиси железа. Приливают 10 - 15 мл раствора едкого натра и нагревают для коагуляции осадка. Раствор с осадком фильтруют через фильтр средней плотности «красная лента», промывают горячей водой.
Осадок растворяют на фильтре 15 - 20 мл горячей разбавленной 1:1 соляной кислотой. Фильтр тщательно промывают горячей водой. Раствор собирают в коническую колбу вместимостью 250 мл.
К горячему раствору добавляют пять капель раствора сульфосалициловой кислоты и уксуснокислый аммоний до рН 2 - 3 и титруют раствором трилона Б до перехода пурпурной окраски в соломенно-желтую. Прибавляют небольшую порцию уксуснокислого аммония. Если наблюдается появление розовой окраски, продолжают титрование до соломенно-желтой окраски раствора.
В случае неполного отделения хрома наблюдается переход окраски от пурпурной до зеленоватой.
4.11.4. Обработка результатов
Содержание железа (Х6) в процентах рассчитывают по формуле
![]()
где Т - титр точно 0,025 М раствора трилона Б по железу, 0,001396 г/мл;
К2- коэффициент молярности раствора трилона Б;
V - объем раствора трилона Б, израсходованный на титрование, мл;
m - масса навески сплава, г.
4.11.1 - 4.11.3. (Измененная редакция, Изм. № 1).
4.12. Определение вольфрама в сплаве РС-1714 гравиметрическим методом
4.12.1. Сущность метода
Метод основан на осаждении вольфрама b-нафтохинолином при рН 1 - 6 после отделения от кремния, хрома и железа.
Коэффициент вариации не должен быть более 1 %.
4.12.2. Материалы, реактивы:
весы лабораторные аналитические типа ВЛА-200г-М;
чашка ПЛ 118-4 по ГОСТ 6563-75;
крышка ПЛ 101-7 по ГОСТ 6563-75;
тигель ПЛ 100-7 по ГОСТ 6563-75;
муфельная электропечь МП-2УМ. Допускается использовать любую муфельную печь с температурой нагрева до 900 °С;
фильтр обеззоленный «белая лента»;
фильтр обеззоленный «синяя лента»;
вода дистиллированная по ГОСТ 6709-72;
калий-натрий углекислый безводный по ГОСТ 4332-76, х. ч., 1 %-ный раствор;
кислота азотная по ГОСТ 44.61-77, х. ч., концентрированная, разбавленная 1:1 и 2 %-ный раствор по объему;
кислота соляная по ГОСТ 3118-77, х. ч.;
кислота фтористоводородная по ГОСТ 10484-78, х. ч.;
метиловый оранжевый ч.д.а., 0,1 %-ный водный раствор; готовят по ГОСТ 4919.1-77;
b-нафтохинолин, ч. д. а., 2 %-ный раствор в 2 %-ной по объему соляной кислоте;
аммиак водный по ГОСТ 3760-79, х. ч., 25 %-ный раствор.
(Измененная редакция, Изм. № 1, 2).
4.12.3. Проведение испытания
Навеску сплава массой около 0,5 г, взвешенную с погрешностью не более 0,0001 г, помещают в платиновую чашку, смачивают 2 мл воды, добавляют 5 мл фтористоводородной кислоты и оставляют на 15 - 20 мин при комнатной температуре. Добавляют 5 мл азотной кислоты и продолжают растворение сплава при умеренном нагревании. Раствор выпаривают до влажных солей, приливают 5 мл азотной кислоты и снова выпаривают до влажных солей. Последнюю операцию повторяют дважды.
Приливают 30 - 50 мл разбавленной 1:1 азотной кислоты, нагревают и содержимое чашки переносят в стакан вместимостью 250 мл. Приставший к стенкам чашки осадок растворяют несколькими каплями аммиака и смывают водой в тот же стакан. Раствор кипятят 15 - 20 мин для полного растворения солей и коагуляции осадка вольфрамовой кислоты. Осадок отфильтровывают на фильтр «синяя лента» с небольшим количеством фильтровальной массы. Осадок количественно, переносят на фильтр с помощью кусочков обеззоленного фильтра, промывают 2 %-ным по объему раствором азотной кислоты и два раза водой.
Фильтр с осадком помещают в платиновый тигель, озоляют и слегка прокаливают в муфельной печи при температуре 600 - 650°С. В тигель с осадком трехокиси вольфрама добавляют 3 г углекислого калия-натрия, накрывают тигель крышкой и сплавляют в муфельной печи при температуре 800 - 850 °С.
Тигель охлаждают, помещают в стакан вместимостью 250 мл, приливают 100 мл горячей воды и кипятят до полного разложения плава. Тигель обмывают горячей водой и удаляют из стакана. Раствор с осадком фильтруют через фильтр «белая лента». Стакан и фильтр промывают горячим раствором углекислого калия-натрия. Фильтрат и промывной раствор собирают в стакан вместимостью 400 мл, нейтрализуют соляной кислотой по метиловому оранжевому, охлаждают и прибавляют при постоянном перемешивании 70 мл раствора β-нафтохинолина. Выдерживают не менее 10 ч. Осадок отфильтровывают на плотный фильтр «синяя лента», промывают холодным 0,5 %-ным раствором b-нафтохинолина в 1 %-ной по объему соляной кислоте.
Фильтр с осадком помещают в прокаленный и взвешенный платиновый тигель, осторожно озоляют и прокаливают в муфельной печи при температуре 600 - 650 °С до постоянной массы. После охлаждения взвешивают полученный остаток трехокиси вольфрама.
4.12.4. Обработка результатов
Содержание вольфрама (Х7) в процентах по массе рассчитывают по формуле
![]()
где 0,793 - коэффициент пересчета с массы трехокиси вольфрама на массу вольфрама;
m2 - масса тигля с осадком трехокиси вольфрама, г;
т1 - масса пустого тигля, г;
m - масса навески материала, г.
4.12.3 - 4.12.4. (Измененная редакция, Изм. № 1).
4.13. Определение содержания кремния
4.13.1. Сущность метода
Кремний определяют в виде двуокиси кремния, выделенной после сплавления пробы с едким натром, разложения плава водой и выпаривания раствора с серной кислотой. Относительная погрешность определения не должна быть более 0,5 %.
4.13.2. Аппаратура, реактивы и растворы:
тигель ПЛ 100-7 по ГОСТ 6563-75;
тигель с крышкой вместимостью 50 мл из никеля любой марки по ГОСТ 6235-73;
вода дистиллированная по ГОСТ 6709-72;
натрия гидроокись по ГОСТ 4328-77, х. ч.;
кислота серная по ГОСТ 4204-77, х. ч.; кислота соляная по ГОСТ 3118-77, х. ч., раствор 1:10;
кислота фтористоводородная по ГОСТ 10484-78, х. ч.;
фильтр обеззоленный «красная лента».
(Измененная редакция, Изм. № 2).
4.13.3. Проведение испытания
Навеску сплава массой 0,2 г взвешивают с погрешностью не более 0,0001 г и помещают в никелевый тигель. Добавляют 1,5 - 2,0 г едкого натра. Тигель закрывают крышкой, ставят в холодную муфельную печь, постепенно повышают температуру до 500 °С и выдерживают при этой температуре 10 мин.
Тигель не открывают до окончания процесса сплавления. По окончании сплавления тигель охлаждают, выщелачивают плав 100 мл холодной воды в стакане вместимостью 300 мл.
В стакан добавляют 5 мл соляной кислоты и нагревают до растворения осадка, затем добавляют 15 мл серной кислоты и выпаривают раствор до появления паров серного ангидрида. Стакан охлаждают, приливают 100 мл горячей воды, содержащей 10 мл концентрированной соляной кислоты, нагревают раствор до растворения солей, не давая кипеть, и фильтруют через фильтр «красная лента». Осадок кремниевой кислоты на фильтре промывают пять-шесть раз горячим раствором соляной кислоты и восемь-десять раз горячей водой.
Фильтрат выпаривают до появления паров серного ангидрида и вторично выделяют кремниевую кислоту, как описано выше, фильтруя через новый фильтр.
Оба фильтра с осадком кремниевой кислоты помещают в платиновый тигель, осторожно озоляют и прокаливают 1 - 2 ч при 1000 - 1200 °С до постоянной массы.
Прокаленный осадок смачивают двумя-тремя каплями воды, добавляют две-три капли серной кислоты и 2 - 3 мл фтористоводородной кислоты. Осторожно выпаривают раствор досуха и снова прокаливают до постоянной массы.
4.13.4. Обработка результатов
Содержание кремния (Х8) в процентах вычисляют по формуле
![]()
где m1 - масса тигля с осадком до обработки фтористоводородной кислотой, г;
m2 - масса тигля с осадком после обработки фтористоводородной кислотой, г;
0,4672 - коэффициент пересчета двуокиси кремния на кремний;
m - масса навески анализируемого сплава, г.
4.14. Определение содержания азота, водорода и кислорода
4.14.1. Сущность метода
Анализируемый материал расплавляют в кварцевой печи с индукционным нагревом графитового тигля в условиях высокого вакуума с применением железо-никель-оловянной «ванны». При этом кислород, как растворенный в металле, так и находящийся в виде окислов, связывается в окись углерода, а водород и азот извлекают из металла в молекулярном состоянии.
Весь экстрагированный газ из образца откачивают диффузионным ртутным насосом и анализируют.
Чувствительность метода 1 · 10-4 % по массе. Относительная погрешность определения не должна быть более 20 %.
4.14.2. Аппаратура, материалы и реактивы:
установка типа «Гиредмет С-911М1», можно использовать эволограф VH-8 фирмы «Хераойс»;
аргон газообразный и жидкий по ГОСТ 10157-79;
тигли графита ос. ч. 7 - 4;
бензол по ГОСТ 5955-75, ч. д. а.;
трубка никелевая диаметром 2,5 - 3,0 мм с толщиной стенки 0,05 мм;
сталь инструментальная углеродистая марки У12А, пруток диаметром 6 - 12 мм по ГОСТ 1435-74;
олово гранулированное по ТУ 6-09-2704-78, ч.;
спирт этиловый ректификованный технический по ГОСТ 18300-72, высший сорт.
(Измененная редакция, Изм. № 1, 2).
4.14.3. Подготовка к испытанию
Анализируемый сплав массой 0,02 - 0,03 г помещают в тонкостенную гильзу длиной 10 - 11 мм, изготовленную из никелевой трубки.
Гильзы, используемые для анализа предварительно прокаливают в вакууме при температуре 1100° - 1200 °С в течение 2 - 3 ч.
В установку загружают гильзы с образцами, перед анализируемыми образцами помещают «железную ванну» (кусочки углеродистой стали), 0,6 - 0,8 г олова и одну - две пустые гильзы.
Отношение веса анализируемого сплава к массе железа ванны должно быть не менее, чем 1:10.
4.14.4. Проведение испытания
Работа на установке проводится в соответствии с инструкцией по эксплуатации.
Вакуумную печь дегазируют при температуре 1850 - 1900 °С до тех пор, пока холостое газовыделение из печи не установится на уровне не более 0,005 см3 за 10 мин. После достижения указанного уровня газовыделения температуру тигля снижают до рабочей, 1650 °С, и сбрасывают в тигель железо и олово, которые дегазируют 15 - 20 мин. Затем в тигель сбрасывают пустую гильзу.
Выделяющийся из гильзы газ откачивают диффузионным ртутным насосом в течение 10 мин, после чего переводят его потоком несущего газа (аргон с добавкой 0,01 % объемных бензола) в хроматограф. Данные, полученные при анализе пустой гильзы, используют как холостую поправку в последующих расчетах.
Анализ гильз с порошком анализируемого сплава проводят так же, как и анализ пустой гильзы.
4.14.5. Калибровка хроматографа
После анализа образцов печь отключают и калибруют хроматограф, для чего в него через градуированный объем выпускают определенное количество порций калибровочного газа до тех пор, пока на хроматограмме величины пиков соответствующих компонентов анализируемого образца и калибровочного газа не будут равны. Отмечают температуру и давление калибровочного газа во время проведения калибровки.
Калибровочный газ представляет собой смесь аргона с около 25 % СО, 12 % Н2, 11 - 12 % N2 и 1,2 - 1,5 % О2 (% объемные).
Для каждого компонента калибровочного газа (СО, Н2, N2) строится калибровочная кривая в координатах:
площадь пика, мм2;
число напусков калибровочной смеси.
По калибровочному графику находят, какому числу напусков калибровочного газа равен пик определяемого компонента от образца.
4.14.6. Обработка результатов
Содержание кислорода (XО2), азота (XN2), водорода (XН2) в процентах в анализируемом порошке сплава находят по формулам:
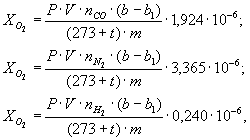
где Р - давление калибровочного газа, выпускаемого в градуированный объем, Па;
V - градуированный объем, см3;
b - число порций калибровочного газа, при котором величины пиков определяемых компонентов от калибровочного и анализируемого газов равны для образца;
b1 - число порций калибровочного газа, при котором величины пиков определяемых компонентов от калибровочного и анализируемого газов равны для пустой гильзы;
nсо, nN2, nH2 - содержание окиси углерода, азота и водорода, соответственно, в калибровочном газе в процентах объемных;
т - масса навески анализируемого порошка, г;
t - температура калибровочного газа, °С;
1,924; 3,365; 0,240 - коэффициенты приведения кислорода, азота и водорода, соответственно, к нормальным условиям и пересчета из объема на граммы.
4.15. Определение содержания углерода
4.15.1. Сущность метода
Определение содержания углерода в сплавах основано на выделении углерода в виде двуокиси при нагревании анализируемого порошка в атмосфере очищенного кислорода.
Свободный углерод определяют при нагреве порошка до 600 °С, а углерод, растворенный в сплаве и связанный в карбид - при сплавлении порошка с окисью свинца при 1100 - 1200 °С.
Количество образующейся при окислении порошка двуокиси углерода измеряют газовым хроматографом.
Абсолютная чувствительность метода при пересчете на углерод составляет 5 · 10-7 г. Относительная погрешность определения не должна быть более 20 %.
4.15.2. Аппаратура, материалы и реактивы:
установка для определения содержания углерода (черт. 2);
Установка для определения содержания углерода
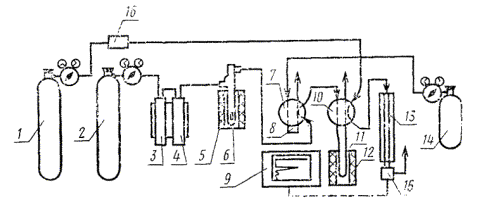
1 - баллон с гелием; 2 - баллон с кислородом; 3, 4 - система очистки кислорода; 5 - электропечь сопротивления на 1100 °С; 6 - окислительная печь; 7, 10 - переключатель газовых потоков; 8 - калиброванная емкость; 9 - самописец; 11 - накопительная ловушка; 12 - электропечь сопротивления на 300 °С; 13 - хроматографическая колонка; 14 - калиброванный газ; 15 - детектор; 16 - регулятор скорости газового потока
весы аналитические 2-го класса типа ВЛА-200 г-М;
цеолит синтетический марки СаА (молекулярное сито 5 А) с размером зерен 0,5 - 1,0 мм;
силикагель технический марки КСМ по ГОСТ 3956-76;
магний хлорнокислый безводный (ангидрон);
аскарит, ч.;
кислород газообразный технический по ГОСТ 5583-78;
азот жидкий по ГОСТ 9292-82;
гелий газообразный;
свинца окись по ГОСТ 9199-77, ч. д. а.;
ампулы из кварца, диаметром 12 мм, длиной 45 мм;
шпатель из платины по ГОСТ 6563-75;
спирт этиловый ректификованный технический по ГОСТ 18300-72, высший сорт.
(Измененная редакция, Изм. № 1, 2).
4.15.3. Подготовка к испытанию
Партию кварцевых ампул (6 - 8 шт.) предварительно очищают прокаливанием в токе кислорода при 1000 - 1100 °С в окислительной печи в течение 1 - 2 ч.
Загружают в прокаленную ампулу навеску окиси свинца массой 0,5 г и прокаливают в кислороде при 700 - 750 °С в течение 3 - 4 ч. Полноту обезуглероживания определяют холостым опытом. Взвешивают ампулу со свежепрокаленной окисью свинца, загружают в нее 0,01 - 0,02 г анализируемого порошка, тщательно перемешивают платиновым шпателем и взвешивают ампулу. При проведении испытания необходимо соблюдать меры, предотвращающие загрязнение ампул и порошка. Брать ампулу чистым пинцетом и хранить между операциями в чистом боксе.
4.15.4. Проведение испытания
Ампулу с образцом и окисью свинца помещают в окислительную печь, проверяют герметичность печи, устанавливают скорость кислорода 50 мл/мин и включают нагрев. Поднимают температуру печи до 600 °С для определения свободного углерода и выдерживают в течение 10 мин. Образующаяся при окислении образца двуокись углерода переносится потоком кислорода в накопительную ловушку 11, содержащую 3 г цеолита (молекулярное сито, обладающее свойством адсорбировать двуокись углерода при комнатной температуре и десорбировать при нагреве до 270 - 300 °С). Выключают нагрев печи и через 2 мин переключатель газовых потоков 10 переводят на поток гелия (50 мл/мин). Электрической печью сопротивления 12 нагревают накопительную ловушку до 300 °С. Выделяющаяся при этом двуокись углерода потоком гелия переносится в хроматограф и регистрируется в течение 6 - 7 мин. Переводят переключатель газовых потоков на поток кислорода и продолжают окисление образца и определение описанным способом, но уже связанного углерода при нагреве печи до 1200 °С.
4.15.5. Калибровка хроматографа
Калибровку хроматографа проводят путем последовательного напуска в накопительную ловушку разных количеств калибровочного газа через переключатель 7, имеющий петлю калибровочного объема (0,1 - 0,5 см3). Калибровочный газ представляет собой известного состава газовую смесь кислорода и двуокиси углерода (около 15 % объемных).
Пик двуокиси углерода от калибровочного газа должен быть равен или чуть больше пика двуокиси углерода от образца.
4.15.6. Обработка результатов
По данным калибровки строят градуировочную кривую в координатах: площадь пиков двуокиси углерода в мм2 - число напусков калибровочного газа.
По этой кривой находят, какому числу напусков калибровочного газа равен пик двуокиси углерода от анализируемого образца. Замеряют температуру и давление калибровочного газа во время проведения анализа.
Содержание углерода (Х9) в процентах вычисляют по формуле
![]()
где Р - атмосферное давление, Па;
Vк - калибровочная емкость, см3;
b2 - число напусков калибровочного газа для образца;
b3 - число напусков калибровочного газа для холостого опыта;
nсо2 - содержание двуокиси углерода в калибровочном газе, % по объему;
t - температура калибровочного газа, °С;
m - масса навески образца, г.
4.16. Определение содержания алюминия и меди в сплаве марки РС-1714.
4.16.1. Сущность метода
Анализ проводят спектральным методом добавок, испаряя пробу с добавками из кратера графитового электрода в дуге постоянного тока. Коэффициент вариации не должен быть более 25 - 30 %.
4.16.2. Аппаратура, материалы и реактивы
спектрограф кварцевый средней дисперсии типа ИСП-30 с однолинзовой системой освещения и трехступенчатым ослабителем;
микрофотометр типа МФ-2 или ИФО;
источник постоянного тока, обеспечивающий напряжение не менее 200 В и рассчитанный на нагрузку до 20 А;
станок для заточки графитовых электродов типа «Универсал» завода «Станкоконструкция»;
весы торсионные типа ВТ-500;
фотопластинки типа I, чувствительность 4,5 ед.;
порошок графитовый ос. ч.;
угли графитированные ос. ч.;
проявитель и фиксаж, приготовленные по ГОСТ 10691.1-73;
натрий хлористый для спектрального анализа, х. ч.;
медь окись по ГОСТ 16539-79, ч. д. а.;
алюминия гидрат окиси по ГОСТ 11841-76, ч. д. а.
4.16.3. Приготовление контрольных образцов
Контрольные образцы, используемые в качестве добавок к пробам, готовят на основе чистого графического порошка (нулевой контрольный образец).
Для приготовления головного контрольного образца, содержащего по 1 % определяемых элементов, навески окислов определяемых элементов (в пересчете на металл) смешивают с навеской графитового порошка.
Контрольные образцы с меньшим содержанием примесей готовят из головного методом последовательного разбавления.
Состав используемых контрольных образцов (добавок) приведен в табл. 2.
|
Концентрация определяемого элемента по массе, % |
||
|
Медь |
Алюминий |
|
|
0 |
- |
- |
|
I |
1 · 10-3 |
2 · 10-3 |
|
II |
1 · 10-2 |
2 · 10-2 |
|
III |
1 · 10-1 |
2 · 10-1 |
При приготовлении контрольных образцов в них вводят 10 % хлористого натрия.
4.16.4. Подготовка к испытанию
Четыре навески анализируемой пробы массами по 200 мг смешивают с навесками массами по 200 мг контрольных образцов 0, I, II, III (добавки) в агатовой ступке, последовательно в порядке возрастания концентрации.
Подготовленные смеси плотно набивают в кратер нижнего графитового электрода - анода (глубина 4,5 мм, диаметр 3,5 мм).
Верхний электрод-катод - графитовый стержень диаметром 6 мм, заточенный на усеченный конус.
4.16.5. Проведение испытания
Испарение смесей проводят в дуге постоянного тока силой 10 А. Расстояние между электродами 2 мм.
Съемку спектров проводят на спектрографе при ширине щели 0,020 мм, экспозиция - 60 с.
Регистрацию спектров осуществляют в области длин волн 230 - 340 нм.
Проявление и фиксирование фотопластинок проводят по ГОСТ 10691.0-84 при температуре 20 ± 1 °С.
Измерение почернений аналитических линий (Sл+ф) и близлежащего фона (Sф) проводят на микрофотометре.
Применяют следующие аналитические линии: меди 327,39 нм; алюминия 308,22 нм.
(Измененная редакция, Изм. № 2).
4.16.6. Обработка результатов
По характеристической кривой фотопластинки находят для всех измеренных почернений (линии вместе с фоном Sл+ф и линии фона Sф) соответствующие им значения lg Iл+ф, lg Iф.
Примечание. Характеристическую кривую строят для данного типа фотопластинок, пользуясь трехступенчатым ослабителем, входящим в комплект спектрографа.
Вычисляют интенсивности, вычитают фон и рассчитывают содержание определяемых элементов (Сх) в процентах по формуле
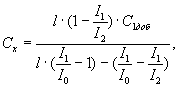
где C1доб - величина вводимой добавки 1, процент по массе;
l - отношение концентрации добавки II к добавке I;
I0, I1, I2 - интенсивности линий элементов в анализируемой пробе с добавками 0, I, II соответственно.
Контрольный образец III используют в том случае, если содержание меди и алюминия в анализируемой пробе выше содержания элементов во втором контрольном образце.
4.17. Определение смачиваемости спиртом
Проверку порошка сплава марки РС-1714 на смачиваемость спиртом проводят путем заливки 3 - 5 г порошка 20 - 25 мл этилового ректификованного спирта и перемешивания. После перемешивания на поверхности не должно быть пленки, не смоченной спиртом.
4.18. Определение содержания кобальта в сплаве РС-5406
4.18.1. Сущность метода
Метод основан на окислении кобальта (II) до кобальта (III) раствором железосинеродистого калия в щелочной среде. Хром III маскируют смесью глицерина и лимонной кислоты.
Относительная погрешность результата анализа должна быть не более 2,5 %.
4.18.2. Аппаратура, материалы, реактивы:
потенциометр любого типа с гальванометром чувствительностью 10-6 А;
мешалка магнитная ММ-3;
электрод индикаторный (+) - проволока ПЛ IM-0,5 по ГОСТ 21007-75;
электрод сравнения (-) - вольфрамовая проволока марки ВА, диаметром 1 мм;
чашка ПЛ 118-4 по ГОСТ 6563-75;
крышка типа «часовое стекло» из фторопласта-4 по ГОСТ 10007-80;
бюретка 1-2-5-0,02 или 1-2-10-0,05 по ГОСТ 20292-74;
пипетка 2-1-25 по ГОСТ 20292-74;
колба 2-100-2 по ГОСТ 1770-74;
стакан В-1-100 ТС по ГОСТ 26336-82;
аммиак водный по ГОСТ 3760-79, ч. д. а., 25 %-ный раствор;
аммоний сернокислый по ГОСТ 3769-78, х. ч., 25 %-ный раствор;
вода дистиллированная по ГОСТ 6709-72;
аммоний лимоннокислый однозамещенный по ГОСТ 7234-79, ч. д. а.;
25 %-ный аммиачный раствор. Готовят следующим образом: 250 г лимоннокислого аммония растворяют в 500 мл воды и добавляют 250 мл аммиака;
калий железосинеродистый, 0,05 н. раствор. Готовят стандарт-титра, хранят в темном месте. 1 мл 0,05 н. раствора соответствует 0,002945 г кобальта;
кислота азотная по ГОСТ 4461-77, х. ч.;
кислота серная по ГОСТ 4204-77, х. ч.;
кислота фтористоводородная по ГОСТ 10484-78, х. ч.;
кислота лимонная по ГОСТ 3652-69, х. ч.;
глицерин по ГОСТ 6259-75, ч. д. а.
(Измененная редакция, Изм. № 2).
4.18.3. Проведение испытания
Навеску образца массой 0,5 г взвешивают с погрешностью ±0,0001 г, помещают в платиновую чашку, добавляют 2 - 3 мл воды, 5 мл фтористоводородной кислоты, накрывают чашку крышкой и осторожно, приподнимая крышку, добавляют по каплям азотную кислоту до полного растворения сплава. Крышку снимают, обмывают водой, в чашку добавляют 2 - 3 мл серной кислоты, осторожно перемешивают раствор и выпаривают его до появления паров серной кислоты. Чашку охлаждают, обмывают стенки 2 - 3 мл воды и снова выпаривают до паров серной кислоты. Содержимое чашки охлаждают, разбавляют водой, переливают в мерную колбу вместимостью 100 мл, доливают до метки водой и перемешивают.
В стакан вместимостью 250 мл наливают 2,5 мл раствора сернокислого аммония, 13,5 мл раствора аммиака, 25 мл глицерина и добавляют 1,5 г лимонной кислоты. Полученный раствор тщательно перемешивают магнитной мешалкой и медленно струей вводят при помощи пипетки 25 мл анализируемого раствора. Стенки стакана обмывают водой, после чего общий объём раствора не должен превышать 100 мл.
В подготовленный таким образом раствор погружают электроды, включают мешалку, переключают потенциометр в положение «титрование» и добавляют раствор железосинеродистого калия из бюретки сначала по 0,2 - 0,5 мл, затем - по 0,1 мл и к концу титрования - по 0,05 мл до резкого скачка потенциала. Эквивалентный объем раствора железосинеродистого калия, израсходованный на титрование кобальта (V1), мл, рассчитывают по формуле
![]()
где V - объем железосинеродистого калия в момент резкого скачка потенциала, мл;
п - разность объемов раствора железосинеродистого калия, прибавляемых в области резкого скачка потенциалов, мл;
E1 - разность потенциалов, предшествующая максимальной разности потенциалов, мВ;
Е2 - разность потенциалов, последующая за максимальной разностью потенциалов, мВ.
4.18.4. Содержание кобальта в навеске образца сплава (X10) в % по массе рассчитывают по формуле
![]()
где т - масса навески сплава, соответствующая аликвотной части раствора, мл.
Результатом анализа считают среднее арифметическое из числа параллельных определений (не менее трех).
4.18 - 4.18.4. (Измененная редакция, Изм. № 1).
5. МАРКИРОВКА, УПАКОВКА, ТРАНСПОРТИРОВАНИЕ И ХРАНЕНИЕ
5.1. Порошки кремниевых сплавов массой от 100 до 200 г упаковывают в двойные пакеты из полиэтиленовой пленки по ГОСТ 10354-82 или пленки ПЭТФ; размеры пакетов определяются массой упаковываемого порошка.
По требованию потребителей допускается фасовка порошков сплавов в пакеты массой 50 г.
Допускается упаковывать порошок в стеклянные или полиэтиленовые банки Б-5п, Бо-5п, Б-6 по ГОСТ 3885-73, при этом масса порошка должна быть от 250 до 400 г.
Фасовку порошка производят с погрешностью не более 1 г.
(Измененная редакция, Изм. № 2).
5.2. Пакеты предварительно протирают бязевой салфеткой, смоченной этиловым ректификованным техническим спиртом высшего сорта по ГОСТ 18300-72.
(Измененная редакция, Изм. № 1).
Банки перед заполнением промывают порошком «Лотос» и протирают бязевой салфеткой, смоченной этиловым ректификованным спиртом.
5.3. После фасовки порошка пакеты заваривают герметичным швом, между внутренним и наружным пакетами предварительно вкладывают этикетку.
При упаковывании в банки для дополнительной герметизации место соединения крышки с банкой должно быть обклеено липкой лентой шириной не менее 20 мм. На банку наклеивают этикетку.
5.4. На этикетках должны быть указаны:
товарный знак предприятия-изготовителя;
наименование и марка сплава;
номер партии;
масса нетто порошка в данной упаковке;
номер фракции порошка;
дата изготовления (месяц, год);
обозначение настоящего стандарта;
штамп отдела технического контроля.
(Измененная редакция, Изм. № 1).
5.5. Пакеты с порошком резистивных кремниевых сплавов помещают в полистироловые коробки. В каждую коробку укладывают пакеты с порошком одной партии.
На коробке должны быть указаны:
товарный знак предприятия-изготовителя;
наименование и марка сплава.
Каждую банку с порошком обертывают гофрированным картоном по ГОСТ 7376-84 в один - два слоя.
(Измененная редакция, Изм. № 2).
5.6. Полистироловые коробки, стеклянные или полиэтиленовые банки укладывают в картонные ящики по ГОСТ 22637-77 или фанерные ящики по ГОСТ 22638-77, выстланные внутри полиэтиленовой пленкой по ГОСТ 10354-73. Банки должны быть переложены гофрированным картоном в горизонтальной и вертикальной плоскостях.
Допускается применять деревянные ящики по ГОСТ 22638-77.
(Измененная редакция, Изм. № 1, 2).
5.7. В каждую полистироловую коробку или ящик с банками вкладывают сертификат, который должен содержать следующие данные:
товарный знак предприятия-изготовителя;
наименование и марку сплава;
номер фракции порошка;
результаты анализов;
номер партии;
дату изготовления (месяц, год);
массу упакованного материала;
отметку отдела технического контроля предприятия-изготовителя;
обозначение настоящего стандарта.
(Измененная редакция, Изм. № 1).
5.8. Маркирование ящиков следует производить по ГОСТ 14192-77.
5.10. Резистивные кремниевые сплавы в упаковке изготовителя хранят в отапливаемых помещениях при температуре окружающей среды от 10 до 40 °С и отсутствии паров кислот и других агрессивных веществ.
5.11. Кремниевые сплавы транспортируют любым видом транспорта при любой температуре.
(Измененная редакция, Изм. № 1).
6. УКАЗАНИЯ ПО ПРИМЕНЕНИЮ
6.1. Все операции с резистивными кремниевыми сплавами для предотвращения загрязнений следует проводить в обеспыленном кондиционируемом помещении при соблюдении правил электронной гигиены.
(Измененная редакция, Изм. № 1).
6.2. Первичную упаковку предприятия-изготовителя следует вскрывать непосредственно перед использованием кремниевых сплавов. Неиспользованный порошок рекомендуется хранить в бюксе или банке с притертой пробкой, не высыпая из пакета, или в эксикаторе с силикагелем в течение установленного гарантийного срока.
6.3. После длительного хранения (более трех месяцев) порошка резистивных кремниевых сплавов рекомендуется просушить в сушильном шкафу при температуре 100 - 120 °C в течение 1 ч с целью улучшения их текучести.
(Измененная редакция, Изм. № 1).
6.4. Дополнительная обработка порошков сплавов перед использованием не требуется. Допускается промывать порошок в чистом этиловом ректификованном спирте по ГОСТ 5962-67 с последующей сушкой по п. 6.3.
7. ГАРАНТИИ ИЗГОТОВИТЕЛЯ
7.1. Изготовитель гарантирует соответствие резистивных кремниевых сплавов требованиям настоящего стандарта при соблюдении потребителем условий хранения, установленных настоящим стандартом.
7.2. Гарантийный срок хранения резистивных кремниевых сплавов - 2 года со дня изготовления.
ПРИЛОЖЕНИЕ 1
Справочное
Физические и электрические параметры резистивных кремниевых сплавов
|
Значения параметров для сплавов марок |
||||||||
|
РС-5406 |
РС-5402 |
РС-4800 |
РС-4400 |
РС-3710 |
РС-3001 |
PC-1714 |
PC-1004 |
|
|
Объемные образцы |
|
|
|
|
|
|
|
|
|
Температура плавления, °С |
1400 ± 15 |
1420 ± 15 |
1550 ± 15 |
1500 ± 15 |
1250 ± 15 |
1350 ± 1,5 |
1570 ± 15 |
1380 ± 15 |
|
Плотность, г/см3 |
- |
- |
4,6 - 4,7 |
- |
4,6 - 5,0 |
3,7 - 4,0 |
5,2 - 5,3 |
3,1 - 3,5 |
|
Удельное электрическое сопротивление, 10-4 Ом·см |
2,5 - 3 |
3,4 - 4,0 |
25 - 135 |
30 - 35 |
5 - 7 |
25 - 35 |
2 - 4 |
40 - 50 |
|
Температурный коэффициент сопротивления в интервале температур 20 - 150 °С, 10-4 град-1 |
6 - 0 |
15 - 18 |
3,5 - 45 |
30 - 50 |
15 - 2,5 |
5 - 15 |
7 - 10 |
8 - 12 |
|
Пленки |
|
|
|
|
|
|
|
|
|
Удельное поверхностное сопротивление, кОм/квадрат |
0,01 - 0,50 |
0,005 - 0,1 |
0,1 - 1,0 |
1,0 - 5,0 |
0,05 - 2,00 |
0,8 - 3,0 |
0,3-0,5 |
3,0-50,0 |
|
Толщина, нм |
36 - 60 |
500 - 3000 |
30 - 100 |
- |
15 - 300 |
20 - 100 |
30 - 50 |
30 - 200 |
|
Температурный коэффициент сопротивления в интервале температур от минус 60 до плюс 125 °С, 10-4 град-1, не более |
+0,5 |
-0,3 |
±2 |
±3 |
±2 |
-1 |
±2 |
-15 |
|
Допустимая мощность рассеяния, Вт/см2, не более |
2 |
2 |
5 |
2 |
5 |
5 |
5 |
5 |
|
Необратимое изменение сопротивления после 1000 ч работы под нагрузкой постоянным током 1 Вт/см2 при окружающей температуре 85 °С, %, не более |
1 |
0,5 |
2 |
- |
1 |
1 |
1 |
2 |
Примечания:
1. Объемные образцы получены при выплавке резистивных кремниевых сплавов.
2. Параметры пленок получены при следующих технологических параметрах их получения методом взрывного вакуумнотермического нанесения на установке типа УВН-2М-2:
остаточное давление в камере
в процессе нанесения, Па, не более...................................... 7 - 10-3
испаритель................................................................................ вольфрамовая лента толщиной 0,2 - 0,3 мм
температура испарителя, °С................................................... 1850 - 1950
метод подачи порошка на испаритель.................................. непрерывный с помощью вибропитателя
расстояние от испарителя до подложки, мм....................... 240
материалы подложки............................................................... ситалл
температура подложки, °С..................................................... 360 ± 10
контроль удельного поверхностного
сопротивления в процессе
нанесения пленки..................................................................... по образцовому сопротивлению
термостабилизирующая обработка
напыленной пленки.................................................................. выдержка в вакууме после нанесения пленки в течение 30 мин при температуре 360 ± 10 °С;
температура подложки в момент напуска воздуха в камеру, °С 300 ± 10
При изменении технологических процессов нанесения пленок параметры пленок могут существенно изменяться. Например, температурный коэффициент сопротивления пленок сплава РС-3710 может иметь положительную или отрицательную величину, необратимые изменения поверхностного сопротивления могут быть уменьшены до 0,1 - 0,01 %.
ПРИЛОЖЕНИЕ 2
Рекомендуемое
Область применения и методы нанесения резистивных кремниевых сплавов
|
Область применения и методы нанесения |
|
|
PC-5406 |
Для получения низкоомных резистивных слоев с широким диапазоном удельного сопротивления и положительным температурным коэффициентом сопротивления порядка 10-5 град-1, обладающих коррозионной стойкостью и способностью к адгезии, методом взрывного испарения с вольфрамового испарителя |
|
PC-5402 |
Для получения низкоомных резистивных слоев с низким температурным коэффициентом сопротивления обладающих коррозионной стойкостью и способностью к адгезии, методом взрывного испарения. Возможно получение низкоомных резистивных слоев с отрицательным коэффициентом сопротивления |
|
РС-4800 |
Для получения резистивных промежуточных слоев, обладающих высокой износостойкостью, коррозионной стойкостью и способностью к адгезии, методом взрывного испарения с вольфрамового испарителя |
|
PC-4400 |
Для получения резистивных слоев, обладающих высокой износостойкостью, коррозионной стойкостью и способностью к адгезии и стабильной работе в течение длительного времени при температуре до 400 °С, методом взрывного испарения с вольфрамового испарителя |
|
РС-3710 |
Для получения резистивных слоев тонкопленочных изделий электронной техники общего и частного назначения методом взрывного испарения с вольфрамового испарителя |
|
PC-3001 |
Для получения резистивных слоев прецизионных тонкопленочных изделий электронной техники частного назначения методом испарения навесок порошка с вольфрамового испарителя |
|
PC-1714 |
Для получения резистивных сулоев тонкопленочных изделий электронной техники общего назначения методом испарения навесок порошка в сухом виде или спиртовой суспензии с вольфрамового испарителя на установке типа УВН-2М-2. |
|
PC-1004 |
Для получения высокоомных резистивных слоев тонкопленочных микросхем частного назначения методом взрывного испарения с вольфрамового испарителя |
Примечание. Для получения резистивных и промежуточных слоев может быть использована в качестве испарителя углеграфитовая ткань, покрытая пирографитом, но при этом свойства пленок будут несколько отличаться от указанных в справочном приложении 1. Применяемость свойств таких пленок должна определяться потребителем.
Изготовление резисторов возможно с помощью масочного метода и фотолитографии.
СОДЕРЖАНИЕ